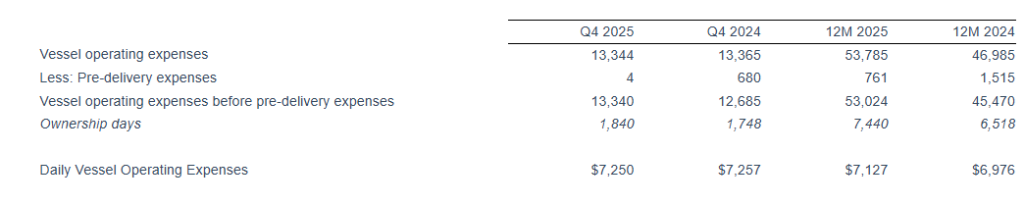FORT WORTH, Texas, Feb. 17, 2026 (GLOBE NEWSWIRE) — Sports Entertainment Gaming Global Corporation (NASDAQ: SEGG, LTRYW) (the “Company” or “SEGG Media”) today announces the successful completion of its previously disclosed acquisition of a controlling interest in Veloce Media Group (“Veloce”), a leading global sports, gaming, and digital media platform.
The acquisition, which values Veloce at approximately $61 million (£45 million), was completed through a blend of cash consideration and SEGG Media equity. The transaction is projected to contribute more than $20 million in additional annual revenue which SEGG Media will begin recognizing and report in the first quarter of this year.
This acquisition marks a significant inflection point in SEGG Media’s strategic transformation into a scaled, revenue-generating global sports, entertainment, and gaming group. Completing on the acquisition of Veloce today demonstrates the current management’s commitment to its shareholders and the wider investor public that the Company is performing on the growth strategy it recently disclosed.
Under the terms of the deal, consideration for the acquisition is a combination of cash and SEGG Media shares priced at $10 per share, highlights the shared belief by SEGG Media’s Board and Veloce’s selling shareholders that SEGG Media’s current share price is grossly undervalued. The structure of the transaction aligns all stakeholders around value creation and sustainable value creation.
Revenue Scale and Valuation Context
Based on reportable incremental revenue of more than $20 million annually from Veloce alone, SEGG Media’s pro forma revenue profile meaningfully grows. Recent market capitalization levels are far below the implied revenue multiple, which should be considered along with the value of the Company’s four domain names and other assets.
Management believes the transaction substantially improves the Company’s revenue-to-market-cap ratio, positioning SEGG Media more comparably with scaled digital media and sports entertainment platforms that trade at materially higher revenue multiples. As Veloce’s operating results are consolidated and reflected in reported financials, the Company will leverage improved scale and operating metrics to provide investors with a clearer framework for valuation assessment.
Daniel Bailey, CEO of Veloce Media Group:“I am delighted to work closely with the wider leadership team to help deliver the next phase of growth. Joining SEGG Media at this pivotal moment is an exciting step for Veloce and our global community. Together, we are building a scaled, future-focused platform with significant opportunity to accelerate growth and deliver long-term value.”
Robert Stubblefield, CFO and Interim CEO and President of SEGG Media: “Closing the Veloce acquisition on schedule is a paradigm shift for SEGG Media. This acquisition strengthens our top line revenue, expands our global footprint, and enhances our ability to drive measurable financial performance for shareholders.”
Strategic Implications for Shareholders
Veloce’s combined platform immediately positions SEGG Media with:
Immediate revenue scale and diversification
Consolidation of operating results from a global digital media asset
Expanded international audience reach
Cross-platform monetization opportunities across Sports.com, Concerts.com, and related assets
A Strengthened balance sheet with increases to assets and equity, and enhanced liquidity.
Management’s immediate focus is on integration execution, maintaining operational discipline, and leveraging revenue scale for continued strong financial performance.
Further updates on integration milestones and strategic operating priorities will be provided in the coming weeks as integration milestones are achieved.
About SEGG Media Corporation SEGG Media (Nasdaq: SEGG, LTRYW) is a global sports, entertainment and gaming group operating a portfolio of digital assets including Sports.com, Concerts.com and Lottery.com. Focused on immersive fan engagement, ethical gaming and AI-driven live experiences, SEGG Media is redefining how global audiences interact with the content they love.
Important Notice Regarding Forward-Looking Statements
This press release contains statements that constitute “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements, other than statements of present or historical fact included in this press release, regarding the Company’s strategy, future operations, prospects, plans and objectives of management, are forward-looking statements. The words “could,” “should,” “will,” “may,” “believe,” “anticipate,” “intend,” “estimate,” “expect,” “project,” “initiatives,” “continue,” the negative of such terms and other similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain such identifying words. These forward-looking statements are based on management’s current expectations and assumptions about future events and are based on currently available information as to the outcome and timing of future events. The forward-looking statements speak only as of the date of this press release or as of the date they are made. The Company cautions you that these forward-looking statements are subject to numerous risks and uncertainties, most of which are difficult to predict and many of which are beyond the control of the Company. In addition, the Company cautions you that the forward-looking statements contained in this press release are subject to risks and uncertainties, including but not limited to, any future findings from ongoing review of the Company’s internal accounting controls, additional examination of the preliminary conclusions of such review, the Company’s ability to secure additional capital resources, the Company’s ability to continue as a going concern, the Company’s ability to respond in a timely and satisfactory matter to the inquiries by Nasdaq, the Company’s ability to regain compliance with the Bid Price Requirement, the Company’s ability to regain compliance with Nasdaq Listing Rules, the Company’s ability to become current with its SEC reports, and those additional risks and uncertainties discussed under the heading “Risk Factors” in the Form 10-K/A filed by the Company with the SEC on April 22, 2025, and the other documents filed, or to be filed, by the Company with the SEC. Additional information concerning these and other factors that may impact the operations and projections discussed herein can be found in the reports that the Company has filed and will file from time to time with the SEC. These SEC filings are available publicly on the SEC’s website at www.sec.gov. Should one or more of the risks or uncertainties described in this press release materialize or should underlying assumptions prove incorrect, actual results and plans could differ materially from those expressed in any forward-looking statements. Except as otherwise required by applicable law, the Company disclaims any duty to update any forward-looking statements, all of which are expressly qualified by the statements in this section, to reflect events or circumstances after the date of this press release.
For additional information, visit http://www.seggmedia.com/ or contact media relations at [email protected].
Declares $0.20 Per Share Dividend and Expands Prompt Newbuilding Program Totaling $226m
Highlights and Developments:
Fifth consecutive year of profitability, delivering adjusted EPS of $1.28, underscoring the resilience and earnings power of Seanergy’s pure-play Capesize strategy across cycles
Declared a Q4 cash dividend of $0.20 per share and total cash dividends for 2025 of $0.43 per share
The Q4 dividend marks the Company’s 17th consecutive quarterly dividend bringing cumulative distributions to $2.64 per share, or approximately $51.2 million
Expanded the prompt newbuilding program to three eco vessels totaling $226million, securing attractive early delivery positions and enhancing future earnings capacity:
Two scrubber-fitted 181,000 dwt Capesize bulkers with expected deliveries in Q2 and Q3 2027
One scrubber-fitted 211,000 dwt Newcastlemax bulker with expected delivery in Q2 2028
Advanced fleet renewal through the sale of the 2010-built M/V Dukeship at a highly attractive valuation, via an 18-month bareboat charter with purchase obligation, generating positive cash flows and releasing significant liquidity
Completed $123.0 million of refinancings at improved terms, generating $51.9 million of incremental liquidity in Q4 and this year to date
Q1 TCE guidance of $25,2732, representing a 14% premium to the average AV5 Baltic Capesize Index year-to-date
____________________________ 1 Adjusted earnings per share, Adjusted Net Income, EBITDA and Adjusted EBITDA are non-GAAP measures. Please see the reconciliation below of Adjusted earnings per share, Adjusted Net Income, EBITDA and Adjusted EBITDA to net income, the most directly comparable U.S. GAAP measure.
ATHENS, Greece, Feb. 17, 2026 (GLOBE NEWSWIRE) — Seanergy Maritime Holdings Corp. (“Seanergy” or the “Company”) (NASDAQ: SHIP), a leading pure-play Capesize shipping company, today reported its financial results for the fourth quarter and twelve months ended December 31, 2025, and announced a quarterly cash dividend of $0.20 per common share. This represents Seanergy’s 17th consecutive quarterly dividend under its capital return policy, with total cash dividends for 2025 of $0.43 per common share, underscoring the Company’s commitment to disciplined capital allocation and consistent shareholder returns.
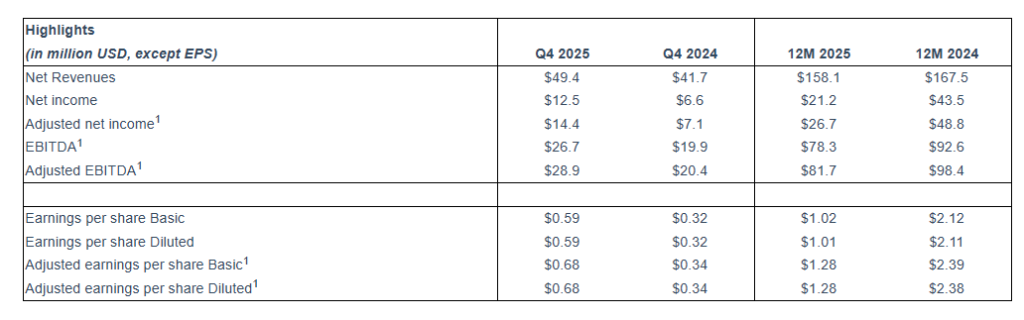
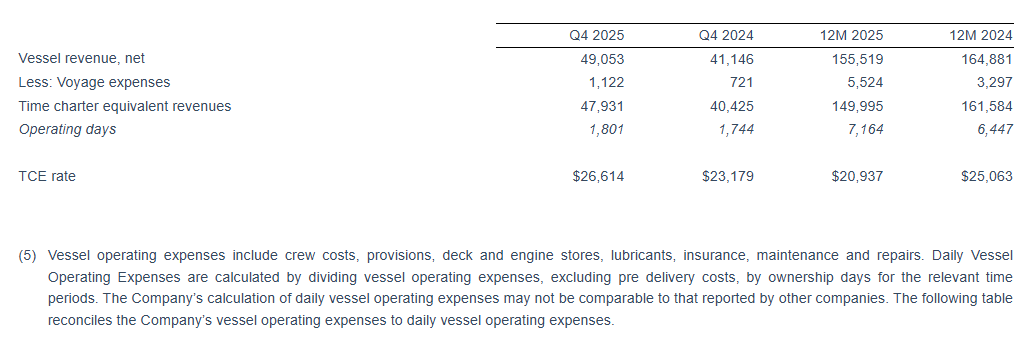
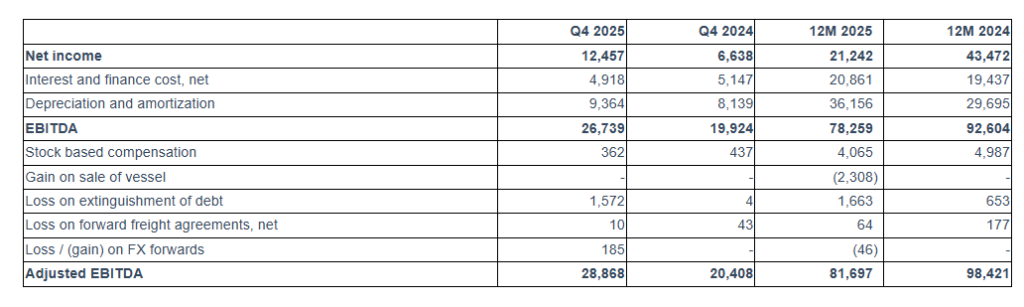
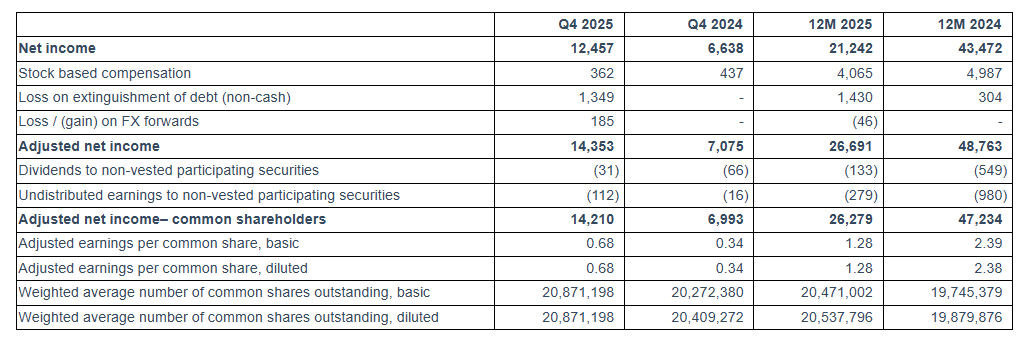
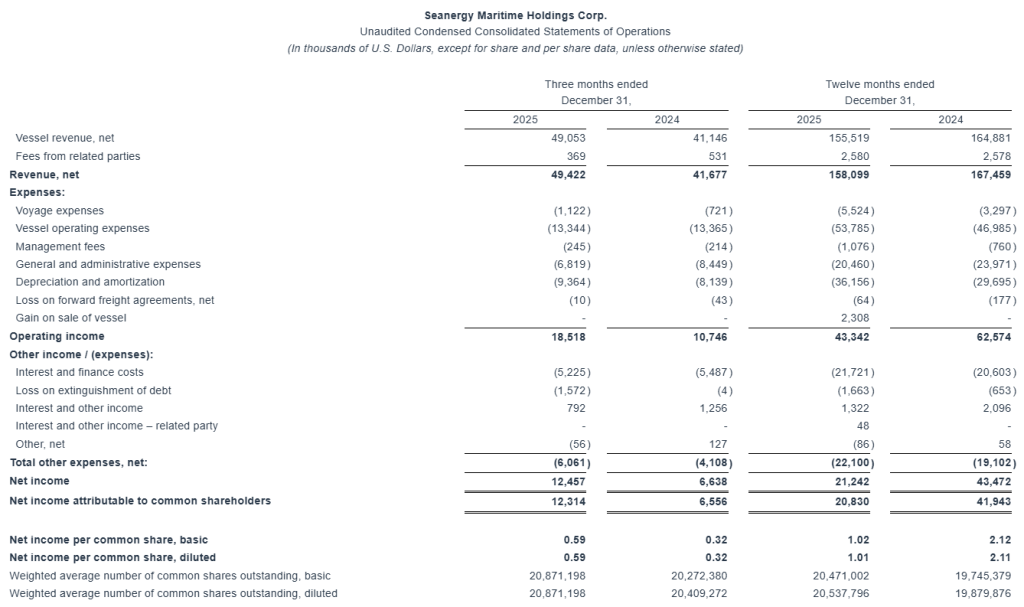
For the quarter ended December 31, 2025, Seanergy generated Net Revenues of $49.4 million, up from $41.7 million in the fourth quarter of 2024. Net Income and Adjusted Net Income for the quarter were $12.5 million and $14.4 million, respectively, compared to Net Income of $6.6 million and Adjusted Net Income of $7.1 million in the fourth quarter of 2024. Adjusted EBITDA for the quarter was $28.9 million, compared to $20.4 million in the same period of 2024. The fleet achieved a daily Time Charter Equivalent (“TCE”) of $26,614 for the fourth quarter of 2025.
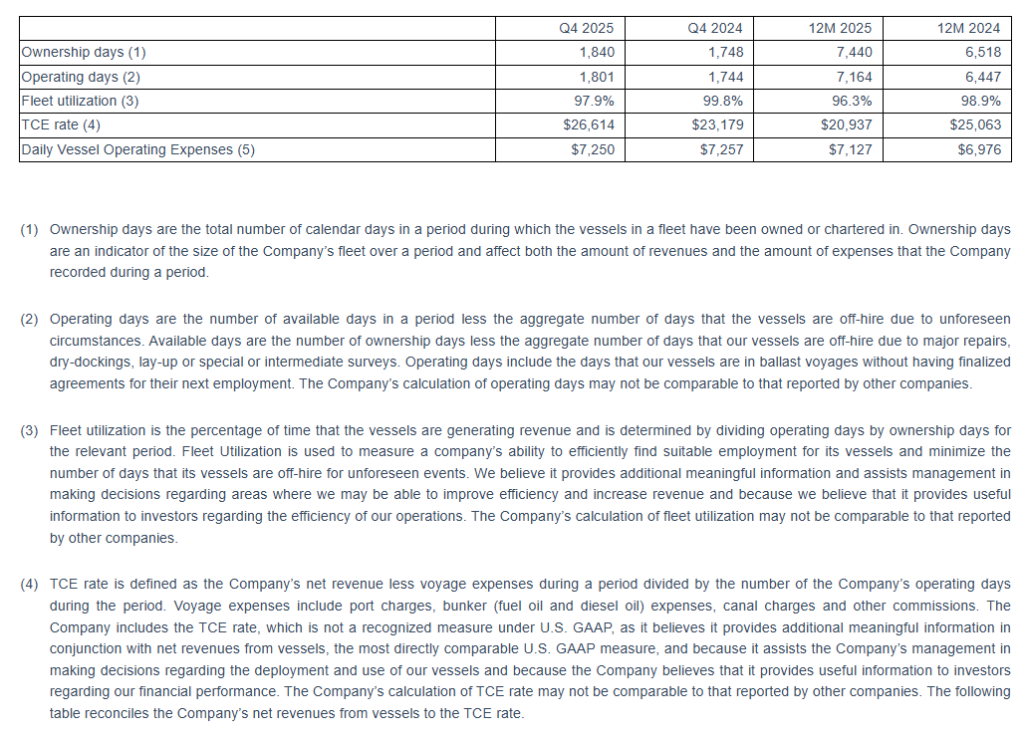
For the full year 2025, Seanergy delivered Net Revenues of $158.1 million, compared to $167.5 million in 2024. Net Income and Adjusted Net Income were $21.2 million and $26.7 million, respectively, compared to Net Income of $43.5 million and Adjusted Net Income of $48.8 million in 2024. Adjusted EBITDA for the twelve months was $81.7 million, compared to $98.4 million for 2024. The daily TCE rate of the fleet for 2025 was $20,937, compared to $25,063 in 2024. The average daily OPEX was $7,127 compared to $6,976 in 2024.
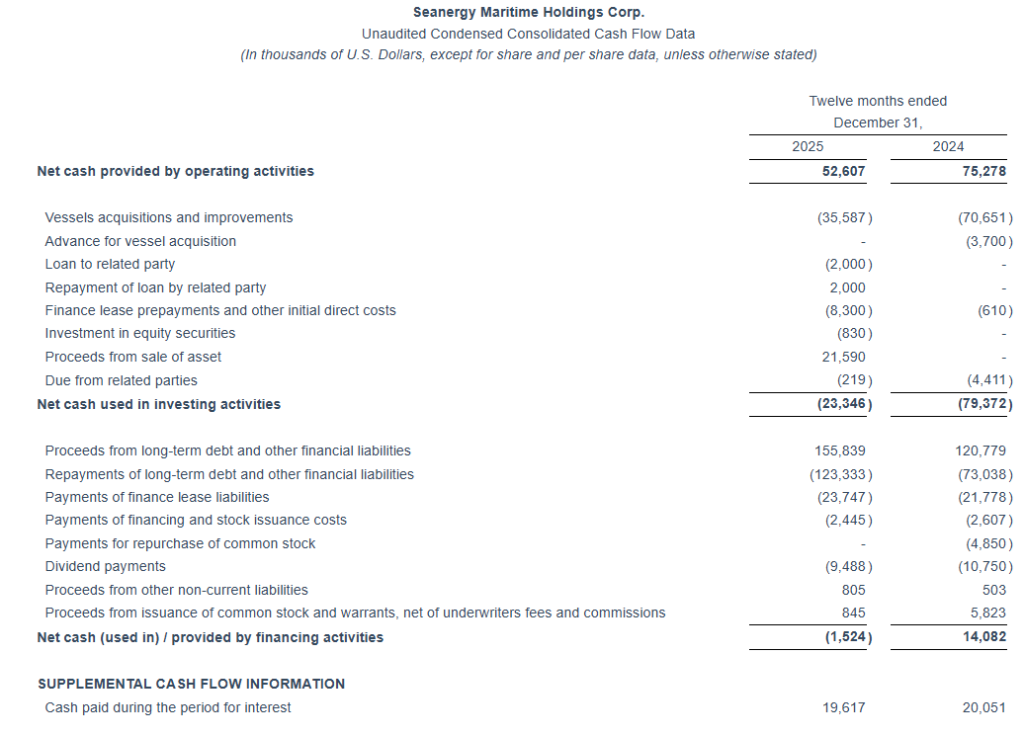
Cash and cash-equivalents and restricted cash, as of December 31, 2025, stood at $62.7 million. Stockholders’ equity at the end of the fourth quarter was $281.4 million. Long-term debt (senior loans and other financial liabilities) net of deferred charges stood at $290.2 million, while the book value of the fleet was $506.7 million, including vessels under construction.
Stamatis Tsantanis, the Company’s Chairman & Chief Executive Officer, stated:
“Driven by a strong Capesize market, Seanergy delivered a very strong fourth quarter, marking our fifth consecutive year of profitability. This performance reflects the durability of our pure-play Capesize strategy, disciplined balance sheet management, and our ability to consistently capture market upside.
“We remain firmly focused on delivering consistent shareholder returns. In 2025, we distributed $0.43 per common share in cash dividends, and with the declaration of the Q4 dividend of $0.20 per common share, we marked our 17th consecutive quarterly dividend. Since launching our dividend program, we have returned $2.64 per common share, or approximately $51.2 million, to our shareholders, underscoring both the strong earnings capacity of our fleet and our disciplined approach to capital allocation.
“Looking ahead, market fundamentals remain constructive as we move into 2026. Robust iron ore and bauxite trade flows, limited Capesize newbuilding supply, and favorable ton-mile dynamics continue to support earnings visibility. With a high-quality fleet, predominantly index-linked employment, and balanced leverage profile, we believe Seanergy is well positioned to capture meaningful upside in this favorable environment.
“Our fleet renewal program is progressing as planned and remains a core strategic priority. In recent months, we added two prompt, eco newbuilding orders at leading Chinese shipyards: a scrubber-fitted Capesize sister vessel to the unit previously announced, scheduled for delivery in Q3 2027, and a scrubber-fitted Newcastlemax scheduled for delivery in Q2 2028. The total current newbuilding investment of approximately $226 million reflects our intention to continue pursuing selective and prompt newbuilding opportunities when market conditions and financing terms are favorably aligned.
“In parallel, and taking advantage of firm secondhand values, we recently agreed to sell the 2010-built Dukeship through an 18-month bareboat arrangement, crystallizing a solid price and generating positive cash flows through the bareboat period. We continue to actively evaluate opportunities to optimize our fleet through selective acquisitions and targeted disposals, while keeping long-term shareholder value and returns as a top priority.
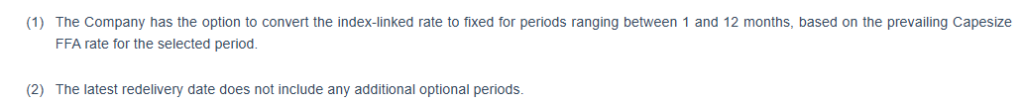
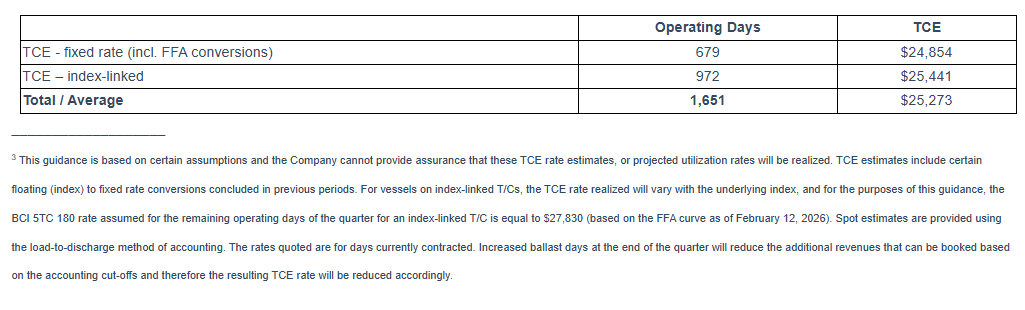
“On the commercial front, we secured index-linked renewals for five vessels, maintaining full participation in a strengthening market while selectively utilizing FFAs to manage volatility. This disciplined approach continues to deliver strong commercial performance. For the first quarter of 2026, we estimate a daily TCE of approximately $25,300, representing a 14% premium to the prevailing AV5 BCI year-to-date, based on the current FFA curve, with approximately 77% of available days fixed at an average rate of $24,739.
“Seanergy enters 2026 from a position of financial strength, operational excellence, and strategic clarity, with a clear path toward continued per-share value creation for our shareholders.”
______________________________ 2 This guidance is based on certain assumptions and the Company cannot provide assurance that these TCE rate estimates, or projected utilization rates will be realized. TCE estimates include certain floating (index) to fixed rate conversions concluded in previous periods. For vessels on index-linked T/Cs, the TCE rate realized will vary with the underlying index, and for the purposes of this guidance, the BCI 5TC 180 rate assumed for the remaining operating days of the quarter for an index-linked T/C is equal to $27,830 (based on the FFA curve as of February 12, 2026). Spot estimates are provided using the load-to-discharge method of accounting. The rates quoted are for days currently contracted. Increased ballast days at the end of the quarter will reduce the additional revenues that can be booked based on the accounting cut-offs and therefore the resulting TCE rate will be reduced accordingly.
Company Fleet:
Fleet Data:
(U.S. Dollars in thousands)
(In thousands of U.S. Dollars, except operating days and TCE rate)
(In thousands of U.S. Dollars, except ownership days and Daily Vessel Operating Expenses)
Net income to EBITDA and Adjusted EBITDA Reconciliation:
(In thousands of U.S. Dollars)
Earnings Before Interest, Taxes, Depreciation and Amortization (“EBITDA”) represents the sum of net income, net interest and finance costs, depreciation and amortization and, if any, income taxes during a period. EBITDA and Adjusted EBITDA are not recognized measurements under U.S. GAAP. Adjusted EBITDA represents EBITDA adjusted to exclude stock-based compensation, gain on sale of vessel, loss on forward freight agreements, net, loss on extinguishment of debt, and loss / (gain) on FX forwards (“Other, net” in statement of operations), which the Company believes are not indicative of the ongoing performance of its core operations.
EBITDA and adjusted EBITDA are presented as we believe that these measures are useful to investors as a widely used means of evaluating operating profitability. Management also uses these non-GAAP financial measures in making financial, operating and planning decisions and in evaluating the Company’s performance. EBITDA and adjusted EBITDA as presented here may not be comparable to similarly titled measures presented by other companies. These non-GAAP measures should not be considered in isolation from, as a substitute for, or superior to, financial measures prepared in accordance with U.S. GAAP.
Adjusted Net Income Reconciliation and calculation of Adjusted Earnings Per Share
(In thousands of U.S. Dollars, except for share and per share data)
To derive Adjusted Earnings Per Share, a non-GAAP financial measure, from Net Income, we adjust for dividends and undistributed earnings to non-vested participating securities and exclude non-cash items, as provided in the table above. We believe that Adjusted Net Income and Adjusted Earnings Per Share assist our management and investors by increasing the comparability of our performance from period to period since each such measure eliminates the effects of such non-cash items as loss on extinguishment of debt, stock based compensation, loss / (gain) on FX forwards and other items which may vary from year to year, for reasons unrelated to overall operating performance. In addition, we believe that the presentation of the respective measure provides investors with supplemental data relating to our results of operations, and therefore, with a more complete understanding of factors affecting our business than with GAAP measures alone. Our method of computing Adjusted Net Income and Adjusted Earnings Per Share may not necessarily be comparable to other similarly titled captions of other companies due to differences in methods of calculation.
First Quarter 2026 TCE Rate Guidance:
As of the date hereof, approximately 77% of the Company fleet’s expected operating days in the first quarter of 2026 have been fixed at an estimated TCE rate of approximately $24,739. Assuming that for the remaining operating days of our index-linked time charters, the BCI 5TC 180 rate will be equal to $27,830 (based on the FFA curve as of February 12, 2026), our estimated TCE rate for the first quarter of 2026 will be approximately $25,2733. The following table provides the breakdown of index-linked charters and fixed-rate charters in the first quarter of 2026:
Fourth Quarter and Recent Developments:
Dividend Distribution for Q3 2025 and Declaration of Q4 2025 Dividend
On January 9, 2026, the Company paid a quarterly cash dividend of $0.13 per common share for the third quarter of 2025 to all shareholders of record as of December 29, 2025.
The Company has declared a quarterly cash dividend of $0.20 per common share for the fourth quarter of 2025 payable on or about April 10, 2026, to all shareholders of record as of March 27, 2026.
Fleet Updates
Newbuilding Contract for a Newcastlemax Vessel at Hantong Shipyard
In November 2025, the Company entered into an agreement for the acquisition of a newbuilding 211,000 dwt scrubber-fitted Newcastlemax vessel from Jiangsu Hantong Ship Heavy Industry Co., Ltd., with delivery expected in the second quarter of 2028. The purchase price is approximately $75.8 million. The first installment, representing 15% of the purchase price, has already been paid. The remaining installments are linked to the vessel’s construction milestones, with 30% of the purchase price payable over the next 2 years and the remaining 55% upon delivery of the vessel.
The new vessel will be built incorporating the latest technological advancements and eco-friendly design features, resulting in enhanced fuel efficiency and reduced emissions in line with the Company’s ongoing fleet renewal and decarbonization strategy.
Newbuilding Contract for a Second Capesize Vessel at Hengli Shipyard
In January 2026, the Company entered into an agreement with Hengli Shipbuilding (Dalian) Co., Ltd. and Hengli Shipbuilding (Singapore) Pte. Ltd. for the construction of a 181,500 dwt scrubber-fitted Capesize vessel. The contract price is approximately $75.2 million, with delivery expected in the third quarter of 2027. The purchase price will be paid in five installments, linked to the vessel’s construction milestones, with 45% of the purchase price payable over the next 14 months and the remaining 55% upon delivery of the vessel.
The new vessel will be built incorporating the latest technological advancements and eco-friendly design features, resulting in enhanced fuel efficiency and reduced emissions in line with the Company’s ongoing fleet renewal and decarbonization strategy.
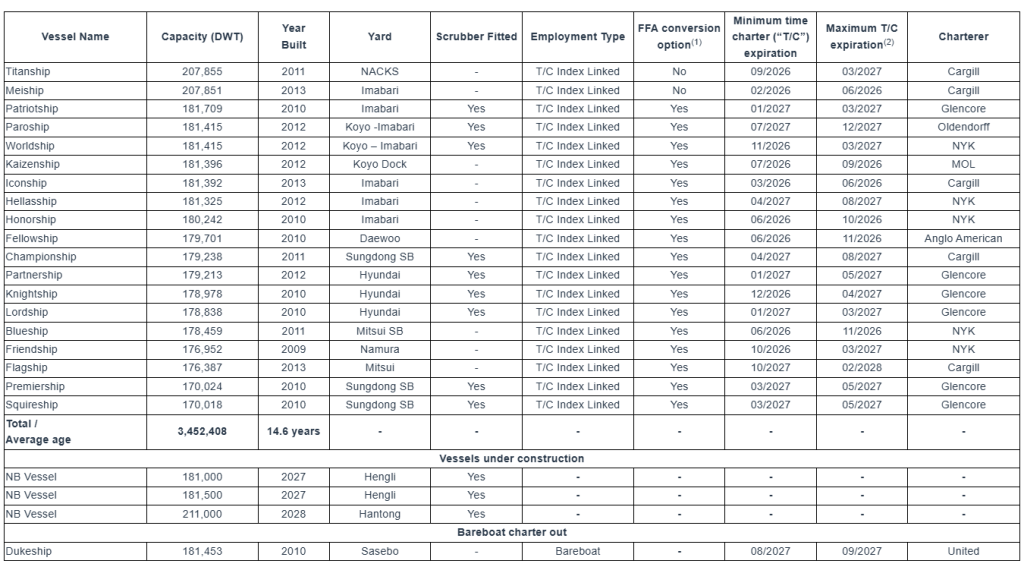
M/V Dukeship – Disposal of Vessel through Bareboat Charter
In February 2026, the Company entered into an agreement with United Maritime Corporation (“United”), a related party, for the disposal of the M/V Dukeship through an 18-month bareboat charter. The charter period commenced following the delivery of the vessel on February 12, 2026. United has advanced a downpayment of $5.5 million and will pay a daily charter rate of $9,450, with a purchase obligation of $22.1 million at the end of the bareboat charter. A special committee of disinterested members of our Board of Directors negotiated the terms and approved the agreement.
Commercial Updates
M/V Flagship – New T/C agreement
In December 2025, the M/V Flagship commenced a new T/C agreement with Cargill International SA with the agreement set to terminate between November 1, 2027 to February 1, 2028, each date subject to (+/- 15 days). The daily hire is based on the 5 T/C routes of the BCI, with an option for the Company to fix the rate for 3 to 9 months based on the prevailing Capesize FFA curve.
M/V Paroship – New T/C agreement
In December 2025, the M/V Paroship commenced a new T/C agreement with Oldendorff GMBH & CO. KG., Ltd for a period of about 20 to about 24 months. The daily hire is based on the 5 T/C routes of the BCI, with an option for the Company to fix the rate for 3 to 9 months based on the prevailing Capesize FFA curve. The Company will also receive most of the benefit from the scrubber profit-sharing scheme.
M/V Friendship – New T/C agreement
In January 2026, the M/V Friendship commenced a new T/C agreement with Glencore Freight Pte. Ltd (“Glencore”) for a period of about 10 to about 14 months. The daily hire is based on the 5 T/C routes of the BCI, with an option for the Company to fix the rate for 1 to 9 months based on the prevailing Capesize FFA curve.
M/V Partnership – New T/C agreement
In February 2026, the M/V Partnership commenced a new T/C agreement with Glencore for a period of about 12 to about 15 months. The daily hire is based on the 5 T/C routes of the BCI, with an option for the Company to fix the rate for 1 to 9 months based on the prevailing Capesize FFA curve. The Company will also receive most of the benefit from the scrubber profit-sharing scheme.
M/V Lordship – Time charter extension
In January 2026, the charterer of the M/V Lordship agreed to extend the time charter agreement in direct continuation from the previous agreement. The extension period will commence on August 21, 2026, for a duration of minimum January 1st, 2027 until maximum March 31st, 2027. The Company receives most of the benefit from the scrubber profit-sharing scheme while the daily hire will be based on a revised premium over the BCI.
M/V Hellasship – Time charter extension
In February 2026, the charterer of the M/V Hellasship agreed to extend the time charter agreement in direct continuation from the previous agreement. The extension period will commence on April 9, 2026, for a duration of minimum 12 to maximum 16 months. The daily hire is based on a revised premium over the BCI, while all other main terms of the time charter remain materially the same.
In December 2025, the Company entered into a new sustainability linked loan facility with Danish Ship Finance secured by the M/Vs Fellowship, Premiership, Championship and Flagship to refinance the sale and leaseback agreement for the M/V Flagship and to increase the existing indebtedness of the other three vessels.
The facility includes a new tranche of $16.8 million secured by the M/V Flagship, with a five-year term. The principal is repayable in 20 quarterly installments of $0.8 million each and a balloon of $1.8 million payable together with the final installment. The interest rate is 2.10% plus 3-month Term SOFR and can fluctuate by 0.05% based on certain emission reduction thresholds.
The additional top-up tranche of $7.3 million, secured by the M/Vs Fellowship, Premiership & Championship, has a three-and-a-half year term and is repayable in 14 quarterly payments of $0.5 million resulting in zero outstanding balance at maturity. The interest rate is 1.95% plus 3-month Term SOFR and can fluctuate by 0.05% based on certain emission reduction thresholds.
M/Vs Hellasship, Patriotship, Iconship & Newbuilding Capesize vessel – Huarong Sale and Leaseback agreements
In December 2025, the Company entered into three separate sale and leaseback agreements totaling $72.5 million for the M/Vs Hellasship, Patriotship & Iconship with entities affiliated with China Huarong Financial Leasing Co., Ltd. The proceeds were used to refinance the outstanding indebtedness of the respective vessels under three sale and leaseback agreements with AVIC International Leasing Co., Ltd. On January 8, 2026, the vessels were sold and chartered back on a bareboat basis for a period of 81 months. The Company has continuous options to purchase the vessels at predetermined prices, starting one year after the commencement date and a purchase obligation at expiry date of each charter. The charterhire principal for the three agreements amortizes in 27 quarterly installments of $2.0 million along with the aggregate purchase obligations of $18.3 million at the expiry of the bareboat charters. Each financing bears interest at a rate of 3-month Term SOFR plus 2.00% per annum, 55 bps lower than the rate of the refinanced agreements. The sale and leaseback agreements do not include any financial covenants or security value maintenance provisions.
Regarding the upcoming delivery of our newbuilding Capesize vessel previously announced, the Company has agreed to enter into a sale and leaseback agreement of $56.3 million to partially finance its acquisition with an entity affiliated with China Huarong Financial Leasing Co., Ltd., which will also provide pre-delivery financing for certain installments under the shipbuilding contract. Upon delivery, the vessel will be sold and chartered back for a period of 60 months. The Company will have continuous purchase options at predetermined prices, commencing one year after the charter commencement date and a purchase obligation at the expiry date. The charterhire principal amortizes in 20 quarterly installments of $0.6 million along with a purchase obligation of $43.5 million at the expiry of the bareboat charter. The financing will bear interest at a rate of 3-month Term SOFR plus 1.80% per annum, while pre-delivery financing amounts will accrue interest payable quarterly in arrears. The sale and leaseback agreement will not include any financial covenants or security value maintenance provisions.
M/V Partnership and Newbuilding Newcastlemax vessel – BOCL Sale and Leaseback agreement
The Company is in the process of finalizing a $26.5 million sale and leaseback agreement for the M/V Partnership with an affiliate of BOC Financial Leasing Corporation Limited to refinance the outstanding indebtedness of the respective vessel under the sale and leaseback agreement with Chugoku Bank, Ltd. The agreement will become effective upon the delivery of the M/V Partnership to the lessor which is expected in March 2026. The Company will sell and charter back the vessel on a bareboat basis for a period of 78 months and will have continuous options to repurchase the vessel at any time following the second anniversary of the delivery at predetermined prices as set forth in the agreement. The charterhire principal will amortize in 26 quarterly installments of $0.8 million along with a purchase option of $6.3 million at the expiry of the bareboat charter. The financing will bear an interest rate of 3-month Term SOFR plus 1.85% per annum, 105 bps lower than the rate of the refinanced agreement. The sale and leaseback agreement will not include any financial covenants or security value maintenance provisions.
Regarding the upcoming delivery of our newbuilding Newcastlemax vessel described above, the Company has agreed to enter into a sale and leaseback agreement of $57.8 million to partially finance its acquisition. The lessor will be an affiliate of BOC Financial Leasing Corporation Limited, which will also provide pre-delivery financing for certain installments under the shipsales contract. Upon delivery, the vessel will be sold and chartered back for a period of 96 months. The Company will have continuous purchase options at predetermined prices as set forth in the agreement, commencing two years after the charter commencement date. The charterhire principal will amortize in 32 quarterly installments of $0.7 million along with a purchase option of $36.3 million at the expiry of the bareboat charter. The financing will bear interest at a rate of 3-month Term SOFR plus 1.85% per annum, while pre-delivery financing amounts will accrue interest payable quarterly in arrears. The sale and leaseback agreement will not include any financial covenants or security value maintenance provisions.
Conference Call:
The Company’s management will host a conference call to discuss financial results on February 17, 2026, at 10:00 a.m. Eastern Time.
Audio Webcast and Earnings Presentation:
There will be a live, and then archived, webcast of the conference call and accompanying presentation available through the Company’s website. To access the presentation and listen to the archived audio file, visit our website, following the Webcast & Presentations section under our Investor Relations page. Participants to the live webcast should register on Seanergy’s website approximately 10 minutes prior to the start of the webcast, following this link.
Conference Call Details:
Participants have the option to register for the call using the following link. You can use any number from the list or add your phone number and let the system call you right away.
About Seanergy Maritime Holdings Corp.
Seanergy Maritime Holdings Corp. is a prominent pure-play Capesize shipping company publicly listed in the U.S. Seanergy provides marine dry bulk transportation services through a modern fleet of Capesize vessels. The Company’s operating fleet consists of 19 vessels (2 Newcastlemax and 17 Capesize) with an average age of approximately 14.6 years and an aggregate cargo carrying capacity of 3,452,408 dwt. Upon the delivery of the newbuilding vessels, the Company’s operating fleet will consist of 22 vessels (3 Newcastlemax and 19 Capesize), with an aggregate cargo carrying capacity of 4,025,908 dwt. Additionally, the Company owns one Capesize vessel that has been chartered out on a bareboat basis.
The Company is incorporated in the Republic of the Marshall Islands and has executive offices in Glyfada, Greece. The Company’s common shares trade on the Nasdaq Capital Market under the symbol “SHIP”.
This press release contains forward-looking statements (as defined in Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended) concerning future events, including with respect to declaration of dividends, market trends and shareholder returns. Words such as “may”, “should”, “expects”, “intends”, “plans”, “believes”, “anticipates”, “hopes”, “estimates” and variations of such words and similar expressions are intended to identify forward-looking statements. These statements involve known and unknown risks and are based upon a number of assumptions and estimates, which are inherently subject to significant uncertainties and contingencies, many of which are beyond the control of the Company. Actual results differ materially from those expressed or implied by such forward-looking statements. Factors that could cause actual results to differ materially include, but are not limited to, the Company’s operating or financial results; the Company’s liquidity, including its ability to service its indebtedness; competitive factors in the market in which the Company operates; shipping industry trends, including charter rates, vessel values and factors affecting vessel supply and demand; future, pending or recent acquisitions and dispositions, business strategy, impacts of litigation, areas of possible expansion or contraction, and expected capital spending or operating expenses; risks associated with operations outside the United States; risks arising from trade disputes between the U.S. and China, including the re-imposition of reciprocal port fees; broader market impacts arising from trade disputes or war (or threatened war) or international hostilities, such as between the U.S. and Venezuela, Israel and Hamas or Iran, China and Taiwan and Russia and Ukraine; risks associated with the length and severity of pandemics; and other factors listed from time to time in the Company’s filings with the SEC, including its most recent annual report on Form 20-F. The Company’s filings can be obtained free of charge on the SEC’s website at www.sec.gov. Except to the extent required by law, the Company expressly disclaims any obligations or undertaking to release publicly any updates or revisions to any forward-looking statements contained herein to reflect any change in the Company’s expectations with respect thereto or any change in events, conditions or circumstances on which any statement is based.
End-to-End Ground System Will Maximize the Full Capabilities of Advanced Software-Defined Satellite
SAN DIEGO, Feb. 17, 2026 (GLOBE NEWSWIRE) — Kratos Defense & Security Solutions, Inc. (Nasdaq: KTOS), a technology company in Defense, National Security and Global Markets, today announced that Airbus Defence and Space has awarded Kratos a contract to deliver a ground segment for its customer Space Communication Technologies (SCT) SPC to support the OmanSat-1 software-defined satellite.
With the growing need for mission flexibility to deliver high throughput services, Oman’s national satellite operator, SCT selected Airbus Defence and Space to deliver OmanSat-1, a fully reconfigurable high-throughput OneSat satellite and the associated ground system. Airbus selected Kratos to deliver the integrated ground system to operate the OmanSat-1 software-defined satellite.
Elodie Viau, Senior Vice President of Telecommunication and Navigation for Airbus Defence and Space, said, “The OmanSat-1 satellite uses a flexible OneSat payload architecture that needs a ground system that is just as dynamic to support the reconfiguration of the satellite and the monitoring of the signals to enable troubleshooting and performance reporting. Building on our successful collaboration across multiple OneSat programs, Kratos will deliver the ground system that works in tandem with the OmanSat-1 satellite, enabling SCT to provide high-throughput services while providing extensive flexibility, serving Oman and the region and expanding to access East African and East Asian markets.”
Reinforcing the need for satellites and ground systems to escape legacy silos and work together in tandem, Luke Wyles, an analyst with Analysys Mason explained in a recent white paper that “in order to optimize the resource allocation on the Software-Defined Satellite (SDS), the network must be orchestrated, reconfiguring the space and ground segment in unison to meet changing demands.”
Bruno Dupas, Vice President of Kratos Space, said, “Kratos is integrating the ground system with Airbus’ mission and control platform to manage and monitor the satellite payload while intelligently orchestrating ground resources. This will enable SCT to dynamically plan spectrum, seamlessly coordinate payload configurations with ground assets, and do so far faster and in a more automated fashion than ever before.”
Kratos will deliver the ground segment that will include the capability to configure and monitor the satellite, optimize and setup the payload using integrated Airbus components, monitor the performance of the entire system from the satellite to the ground and orchestrate part of the operations. The implementation will include Ka-Band telemetry, tracking and command (TT&C) antennas, monitor and control and carrier monitoring software, command, control and flight dynamics capabilities and the orchestration system. Leveraging its office in Oman, Kratos will also perform site surveys, install and commission equipment, deliver training and provide support for the delivery.
Kratos has a proven track record of delivering state-of-the-art ground solutions and is at the forefront of developing new capabilities to support today’s software-defined satellites. This includes the recent successful completion of the first factory acceptance test of Kratos’ Epoch command and control (C2) system with Airbus’ OneSat software-defined satellite. Kratos also brings a breadth of experience across the ground segment in areas such as satellite C2; TT&C; payload operations; network management and spectrum monitoring to deliver integrated satellite and ground system solutions.
About Kratos Defense & Security Solutions Kratos Defense & Security Solutions, Inc. (NASDAQ: KTOS) is a technology, products, system and software company addressing the defense, national security, and commercial markets. Kratos makes true internally funded research, development, capital and other investments, to rapidly develop, produce and field solutions that address our customers’ mission critical needs and requirements. At Kratos, affordability is a technology, and we seek to utilize proven, leading edge approaches and technology, not unproven bleeding edge approaches or technology, with Kratos’ approach designed to reduce cost, schedule and risk, enabling us to be first to market with cost effective solutions. We believe that Kratos is known as an innovative disruptive change agent in the industry, a company that is an expert in designing products and systems up front for successful rapid, large quantity, low-cost future manufacturing which is a value add competitive differentiator for our large traditional prime system integrator partners and also to our government and commercial customers. Kratos intends to pursue program and contract opportunities as the prime or lead contractor when we believe that our probability of win (PWin) is high and any investment required by Kratos is within our capital resource comfort level. We intend to partner and team with a large, traditional system integrator when our assessment of PWin is greater or required investment is beyond Kratos’ comfort level. Kratos’ primary business areas include virtualized ground systems for satellites and space vehicles including software for command & control (C2) and telemetry, tracking and control (TT&C), jet powered unmanned aerial drone systems, advanced vehicles and rocket systems, propulsion systems for drones, missiles, loitering munitions, supersonic systems, space craft and launch systems, C5ISR and microwave electronic products for missile, radar, missile defense, space, satellite, counter UAS, directed energy, communication and other systems, and virtual & augmented reality training systems for the warfighter. For more information, visit www.KratosDefense.com.
Notice Regarding Forward-Looking Statements Certain statements in this press release may constitute “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements are made on the basis of the current beliefs, expectations and assumptions of the management of Kratos and are subject to significant risks and uncertainty. Investors are cautioned not to place undue reliance on any such forward-looking statements. All such forward-looking statements speak only as of the date they are made, and Kratos undertakes no obligation to update or revise these statements, whether as a result of new information, future events or otherwise. Although Kratos believes that the expectations reflected in these forward-looking statements are reasonable, these statements involve many risks and uncertainties that may cause actual results to differ materially from what may be expressed or implied in these forward-looking statements. For a further discussion of risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of Kratos in general, see the risk disclosures in the Annual Report on Form 10-K of Kratos for the year ended December 29, 2024, and in subsequent reports on Forms 10-Q and 8-K and other filings made with the SEC by Kratos.
Alan Smith, Vice President and General Manager of Graham Manufacturing to retire in April 2026 and will serve in an advisory role moving forward
William Zmyndak is expected to assume the role of Vice President and General Manager of Graham Manufacturing upon Mr. Smith’s retirement
Additionally, the Company announces the appointments of Keith Oufnac as Chief Information Officer and Rachel Jaakkola as Chief Human Resources Officer
BATAVIA, N.Y.–(BUSINESS WIRE)– Graham Corporation (NYSE: GHM) (“GHM” or the “Company”), a global leader in the design and manufacture of mission critical fluid, power, heat transfer vacuum, and advanced mixing technologies for the Defense, Energy & Process and Space industries, today announced the appointment of William Zmyndak, Deputy General Manager of Graham Manufacturing.
As part of a proactive succession plan, Alan Smith, currently Vice President and General Manager of Graham Manufacturing, will transition to a consulting and advisory role beginning in April 2026. In this capacity, Mr. Smith will continue to support the business and leadership team through a transition period upon his retirement. Effective April 2026, Mr. Zmyndak will assume the role of Vice President and General Manager of Graham Manufacturing upon Alan’s retirement.
Mr. Zmyndak brings more than three decades of manufacturing and operational leadership experience, including senior leadership and P&L responsibility across complex, multi-site aerospace and industrial businesses. Prior to joining Graham, he served as Vice President and General Manager at ITT Control Technologies, where he led operations across multiple U.S. and international locations and drove margin expansion, operational excellence, and growth initiatives. Earlier in his career, Mr. Zmyndak held senior leadership roles at Kaman Aerosystems, Chromalloy, Barnes Aerospace, and Pratt & Whitney. He holds a Master of Business Administration from Purdue University and a Bachelor of Science in Manufacturing Engineering from Boston University.
In addition to this leadership transition, the Company announced two key leadership appointments that further strengthen its executive team.
Keith Oufnac has been appointed Chief Information Officer. Mr. Oufnac has more than 20 years of experience leading digital transformation, IT strategy, and cybersecurity initiatives across defense, aerospace, and highly regulated industries. Most recently, he served as Vice President of Information Technology at Bollinger Shipyards, where he led enterprise-wide infrastructure modernization, cybersecurity enhancements, and large-scale systems integration efforts, including support for significant acquisition activity.
Rachel Jaakkola has been appointed Chief Human Resources Officer. Ms. Jaakkola is a seasoned human resources executive with over a decade of experience building and scaling people organizations within aerospace, defense, and energy companies. She has a proven track record in talent strategy, leadership development, employee engagement, and M&A integration. Prior to joining Graham, Ms. Jaakkola served in senior HR leadership roles at Barber-Nichols, where she established and led the human resources function through periods of significant growth and organizational transformation.
Matthew J. Malone, President and Chief Executive Officer of Graham Corporation, said, “Alan has been instrumental in strengthening Graham Manufacturing for over 30 years of his career, and we are grateful for his continued support during the transition. Will brings extensive operational and P&L leadership experience across complex manufacturing environments, along with a strong commitment to people and execution. I am confident he is the right leader to build on our momentum and continue driving operational excellence and growth. The additions of Keith and Rachel further strengthen our leadership team as we invest in the systems, people and capabilities needed to support our long-term strategy.”
About Graham Corporation
Graham is a global leader in the design and manufacture of mission-critical fluid, power, heat transfer, vacuum, and advanced mixing technologies for the Defense, Energy & Process, and Space industries. Graham Corporation and its family of global brands are built upon world-renowned engineering expertise, proprietary technologies, as well as its responsive and flexible service and the unsurpassed quality customers have come to expect from the Company’s products and systems. Graham Corporation routinely posts news and other important information on its website, grahamcorp.com, where additional information on Graham Corporation and its businesses can be found.
Safe Harbor Regarding Forward Looking Statements
This news release contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended.
Forward-looking statements are subject to risks, uncertainties and assumptions and are identified by words such as “continue,” “expects,” “will,” “plan” and other similar words. All statements addressing operating performance, events, or developments that Graham Corporation expects or anticipates will occur in the future, including but not limited to, expected future management personnel changes and the timing of such changes, expected expansion and growth opportunities, and its growth strategy, are forward-looking statements. Because they are forward-looking, they should be evaluated in light of important risk factors and uncertainties. These risk factors and uncertainties are more fully described in Graham Corporation’s most recent Annual Report filed with the Securities and Exchange Commission (the “SEC”), included under the heading entitled “Risk Factors”, and in other reports filed with the SEC.
Should one or more of these risks or uncertainties materialize or should any of Graham Corporation’s underlying assumptions prove incorrect, actual results may vary materially from those currently anticipated. In addition, undue reliance should not be placed on Graham Corporation’s forward-looking statements. Except as required by law, Graham Corporation disclaims any obligation to update or publicly announce any revisions to any of the forward-looking statements contained in this news release.
ATLANTA, GA, February 13, 2026 – GeoVax Labs, Inc. (Nasdaq: GOVX) (the “Company”), a clinical-stage biotechnology company developing immunotherapies and vaccines against cancer and infectious diseases, today announced that it has entered into definitive agreements for the purchase and sale of 432,902 shares of its common stock (or pre-funded warrants in lieu thereof) at a purchase price of $2.31 per share (or pre-funded warrant in lieu thereof) in a registered direct offering priced at-the-market under Nasdaq rules (the “Offering”). In a concurrent private placement, the Company will issue unregistered series A-1 warrants to purchase up to 432,902 shares of common stock and unregistered series A-2 warrants to purchase up to 432,902 shares of common stock. The warrants will have an exercise price of $2.31 per share and will be exercisable beginning on the effective date of shareholder approval of the issuance of the shares of common stock upon exercise of the warrants. The series A-1 warrants will expire five years after the date of shareholder approval and the series A-2 warrants will expire two years after the date of shareholder approval.
The closing of the Offering is expected to occur on or about February 17, 2026, subject to the satisfaction of customary closing conditions. The gross proceeds from the Offering are expected to be approximately $1 million, before deducting placement agent fees and other estimated offering expenses. The Company intends to use the net proceeds from the Offering to advance its product candidates, including research and development, manufacturing, clinical studies, and working capital.
H.C. Wainwright & Co. is acting as the exclusive placement agent for the Offering.
The shares (or pre-funded warrants) (but not the unregistered warrants and the shares of common stock underlying the unregistered warrants) in the registered direct offering described above are being offered by the Company pursuant to a shelf registration statement on Form S-3 (File No. 333-277585) previously filed with the Securities and Exchange Commission (the ”SEC”) and declared effective by the SEC on March 13, 2024. The registered direct offering is being made only by means of a prospectus, including a prospectus supplement, forming a part of the effective registration statement, relating to the registered direct offering that will be filed with the SEC. Electronic copies of the final prospectus supplement and accompanying prospectus may be obtained, when available, on the SEC’s website at http://www.sec.gov or by contacting H.C. Wainwright & Co., LLC at 430 Park Avenue, 3rd Floor, New York, New York 10022, by phone at (212) 856-5711 or e-mail at [email protected].
The unregistered warrants described above are being offered in a private placement under Section 4(a)(2) of the Securities Act of 1933, as amended (the “Securities Act”), and/or Regulation D promulgated thereunder and, along with the shares of common stock underlying such unregistered warrants, have not been registered under the Securities Act, or applicable state securities laws. Accordingly, the unregistered warrants and underlying shares of common stock may not be offered or sold in the United States except pursuant to an effective registration statement or an applicable exemption from the registration requirements of the Securities Act and such applicable state securities laws.
The Company also has agreed to amend certain existing warrants to purchase up to an aggregate of 236,000 shares of the Company’s common stock that were previously issued to the investors in July 2025, with an exercise price of $4.35 per share, respectively, effective upon the closing of the offering, such that the amended warrants will have a reduced exercise price of $2.31 per share and will be exercisable beginning on the effective date of shareholder approval of the issuance of the shares upon exercise of the warrants.
This press release shall not constitute an offer to sell or a solicitation of an offer to buy these securities, nor shall there be any sale of these securities in any state or other jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or other jurisdiction.
About GeoVax
GeoVax Labs, Inc. is a clinical-stage biotechnology company developing novel vaccines against infectious diseases and therapies for solid tumor cancers. The Company’s lead clinical program is GEO-CM04S1, a next-generation COVID-19 vaccine currently in three Phase 2 clinical trials, being evaluated as (1) a primary vaccine for immunocompromised patients such as those suffering from hematologic cancers and other patient populations for whom the current authorized COVID-19 vaccines are insufficient, (2) a booster vaccine in patients with chronic lymphocytic leukemia (CLL) and (3) a more robust, durable COVID-19 booster among healthy patients who previously received the mRNA vaccines. In oncology the lead clinical program is evaluating a novel oncolytic solid tumor gene-directed therapy, Gedeptin®, having recently completed a multicenter Phase 1/2 clinical trial for advanced head and neck cancers. GeoVax is also developing a vaccine targeting Mpox and smallpox and, based on recent EMA regulatory guidance, anticipates progressing directly to a Phase 3 clinical evaluation, omitting Phase 1 and Phase 2 trials. GeoVax has a strong IP portfolio in support of its technologies and product candidates, holding worldwide rights for its technologies and products. For more information about the current status of our clinical trials and other updates, visit our website: www.geovax.com.
Forward-Looking Statements
This release contains forward-looking statements regarding GeoVax’s business plans, including, but not limited to, statements regarding the completion of the offering, the satisfaction of customary closing conditions related to the offering, the receipt of shareholder approval and the anticipated use of proceeds from the offering. The words “believe,” “look forward to,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “could,” “target,” “potential,” “is likely,” “will,” “expect” and similar expressions, as they relate to us, are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. Actual results may differ materially from those included in these statements due to a variety of factors, including whether: GeoVax is able to obtain acceptable results from ongoing or future clinical trials of its investigational products, GeoVax’s immuno-oncology products and preventative vaccines can provoke the desired responses, and those products or vaccines can be used effectively, GeoVax’s viral vector technology adequately amplifies immune responses to cancer antigens, GeoVax can develop and manufacture its immuno-oncology products and preventative vaccines with the desired characteristics in a timely manner, GeoVax’s immuno-oncology products and preventative vaccines will be safe for human use, GeoVax’s vaccines will effectively prevent targeted infections in humans, GeoVax’s immuno-oncology products and preventative vaccines will receive regulatory approvals necessary to be licensed and marketed, GeoVax raises required capital to complete development, there is development of competitive products that may be more effective or easier to use than GeoVax’s products, GeoVax will be able to enter into favorable manufacturing and distribution agreements, and other factors, over which GeoVax has no control.
Further information on our risk factors is contained in our periodic reports on Form 10-Q and Form 10-K that we have filed and will file with the SEC. Any forward-looking statement made by us herein speaks only as of the date on which it is made. Factors or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of them. We undertake no obligation to publicly update any forward-looking statement, whether as a result of new information, future developments or otherwise, except as may be required by law.
Transaction Closing Date Set for Next Tuesday, February 17
FORT WORTH, Texas, Feb. 13, 2026 (GLOBE NEWSWIRE) — Sports Entertainment Gaming Global Corporation (NASDAQ: SEGG, LTRYW) (the “Company” or “SEGG Media”), the global sports, entertainment, and gaming group, today announced that it has agreed to binding terms to acquire at least a majority interest in Veloce Media Group (“Veloce”), one of the fastest-growing and market leading platforms operating at the intersection of sport, gaming and digital media.
The completion date for consummating the acquisition is set for Tuesday, February 17, 2026, which will result in SEGG Media acquiring a controlling interest of Veloce, enabling consolidation for accounting and reporting purposes and direct control. The transaction values Veloce at approximately $61 million (£45 million) and is projected to contribute in excess of $20 million in additional annual revenue which will begin to be reported in the first quarter of 2026. SEGG Media’s management views Veloce as a foundational international platform that aligns with the Company’s strategy of acquiring cash-generative, media-driven sports assets capable of scaling across sponsorship, content, and commerce.
The acquisition of Veloce will be completed through a blend of cash consideration and SEGG Media common shares priced at $10 per share. The Veloce acquisition is one that the Company has been hyper-focused on for months and completing the transaction is a paradigm shift for SEGG Media and its shareholders. The targeted acquisition of Veloce by SEGG Media signals the Company’s rapid evolution into a diversified global sports and media group.
Veloce’s recent acquisition of Quadrant, co-founded by the current Formula 1 Champion Lando Norris, is a significant and rapidly growing gaming and lifestyle company. With a portfolio of blue-chip commercial partners and direct revenue generation in apparel and product sales the Quadrant business will continue to play a key role in the revenue growth of Veloce and SEGG Media.
Darryl Eales, Veloce Director and investor and formerly CEO of Lloyd’s Development Capital, commented:“I’m truly excited by the potential of the Veloce and SEGG partnership. High-quality, driven, and aligned management teams are crucial for the delivery of strong shareholder value creation. The combined leadership creates a powerful platform for significant and rapid growth, underpinned by both SEGG’s exciting brands and well-founded sports and entertainment strategy and Veloce’s multi-stream revenue platform and strong financial performance.
“Both the Veloce team and the SEGG Board have remained relentless in executing the transaction – even as SEGG completed the final stages of its turnaround – driven by a combined belief in the significant scale of the opportunity that exists post-completion. With the combined value of Veloce, SEGG, and additional pipeline acquisitions, receiving consideration in $10 SEGG stock represents significant upside for Veloce shareholders.”
Daniel Bailey, CEO of Veloce Media Group,said: “This acquisition represents a defining moment not only for Veloce, but for SEGG Media as a group. From the outset, it was clear that our businesses share a common vision for building a global, digitally led sports media platform with ambition and long-term commercial strength.
“The combination of SEGG Media’s access to public markets and strategic focus with Veloce’s brands, partnerships and proven revenue model creates a powerful foundation for accelerated expansion.”
Veloce’s ecosystem spans championship-winning esports teams, athlete-led content platforms, sustainable motorsport series, and a commercial portfolio supported by global brands including McLaren, Revolut, VISA, LEGO, Microsoft, Hilton, E.ON, and Thrustmaster.
Driving over 500 million views per month, Veloce brings with it rapidly growing and diversified revenue streams across digital content, esports, motorsport and brand partnerships, reporting $17.5 million (£12.8 million) in revenue for its latest reported financial period.
Since the start of 2026, SEGG Media’s strategy has been firmly focused on executing fundamental acquisitions designed to accelerate its growth by establishing a scalable and profitable revenue-generating platform. The integration of Veloce’s business and revenue positions SEGG Media to capitalize on accelerating global demand across sport, media, gaming and digital entertainment, with a clear focus on creating genuine value to the Company driven by consistently improving return on invested capital (ROIC) and sustaining high-quality revenue growth with higher profit margins.
Robert Stubblefield, CFO and Interim CEO and President of SEGG Media, said:“The acquisition of Veloce Media Group is a pivotal acquisition for the Company and a clear validation of the strategic direction we set at the start of 2026. Veloce delivers scale, rapidly growing revenues and high-quality commercial partnerships that materially strengthen our profile.
“This acquisition of Veloce and its subsidiary Quadrant springboards SEGG Media to immediately unlocking significant revenue for the Company, which creates long-term shareholder value especially as we integrate a best-in-class digital sports and media platform into the Company. Simply put, it’s a gamechanger!”
Closing is subject to final legal review, completion of definitive documentation, and customary closing conditions.
About SEGG Media Corporation SEGG Media (Nasdaq: SEGG, LTRYW) is a global sports, entertainment and gaming group operating a portfolio of digital assets including Sports.com, Concerts.com and Lottery.com. Focused on immersive fan engagement, ethical gaming and AI-driven live experiences, SEGG Media is redefining how global audiences interact with the content they love.
Important Notice Regarding Forward-Looking Statements
This press release contains statements that constitute “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements, other than statements of present or historical fact included in this press release, regarding the Company’s strategy, future operations, prospects, plans and objectives of management, are forward-looking statements. When used in this Form 8-K, the words “could,” “should,” “will,” “may,” “believe,” “anticipate,” “intend,” “estimate,” “expect,” “project,” “initiatives,” “continue,” the negative of such terms and other similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain such identifying words. These forward-looking statements are based on management’s current expectations and assumptions about future events and are based on currently available information as to the outcome and timing of future events. The forward-looking statements speak only as of the date of this press release or as of the date they are made. The Company cautions you that these forward-looking statements are subject to numerous risks and uncertainties, most of which are difficult to predict and many of which are beyond the control of the Company. In addition, the Company cautions you that the forward-looking statements contained in this press release are subject to risks and uncertainties, including but not limited to, any future findings from ongoing review of the Company’s internal accounting controls, additional examination of the preliminary conclusions of such review, the Company’s ability to secure additional capital resources, the Company’s ability to continue as a going concern, the Company’s ability to respond in a timely and satisfactory matter to the inquiries by Nasdaq, the Company’s ability to regain compliance with the Bid Price Requirement, the Company’s ability to regain compliance with Nasdaq Listing Rules, the Company’s ability to become current with its SEC reports, and those additional risks and uncertainties discussed under the heading “Risk Factors” in the Form 10-K/A filed by the Company with the SEC on April 22, 2025, and the other documents filed, or to be filed, by the Company with the SEC. Additional information concerning these and other factors that may impact the operations and projections discussed herein can be found in the reports that the Company has filed and will file from time to time with the SEC. These SEC filings are available publicly on the SEC’s website at www.sec.gov. Should one or more of the risks or uncertainties described in this press release materialize or should underlying assumptions prove incorrect, actual results and plans could differ materially from those expressed in any forward-looking statements. Except as otherwise required by applicable law, the Company disclaims any duty to update any forward-looking statements, all of which are expressly qualified by the statements in this section, to reflect events or circumstances after the date of this press release.
Alabama becomes the first Conduent-supported state – and only the second state in the nation – to roll out chip-enabled EBT cards statewide
Chips allow beneficiaries to insert their cards into point-of-sale terminals, significantly enhancing the security of SNAP and TANF accounts
FLORHAM PARK, N.J. — Conduent Incorporated (Nasdaq: CNDT), a global technology-driven business solutions and services company, today announced its collaboration with the Alabama Department of Human Resources (DHR) to introduce chip-enabled EBT cards designed to help prevent fraud. The new cards, now mailed to EBT cardholders across the state, are expected to significantly enhance account security for beneficiaries, including those in the Supplemental Nutrition Assistance Program (SNAP) and the Temporary Assistance for Needy Families (TANF) program.
Across the country, states have reported a sharp rise in fraud attempts targeting EBT cards, which traditionally rely on magnetic stripes and are vulnerable to skimming – where criminals install devices on point-of-sale terminals to steal card information. With chip technology, Alabama cardholders can now insert their cards into the terminals rather than swiping them, adding a critical layer of protection.
Following a pilot program launched in December, Alabama is the first Conduent-supported state – and only the second state nationwide – to introduce EBT cards to all cardholders. Additional states are preparing similar rollouts.
“I am so pleased to finally bring this instrumental change to our EBT cardholders statewide,” said Alabama DHR Commissioner Nancy Buckner. “After a successful pilot program, we have shown that these new cards are easy to use and offer much better protection for the benefits. I am pleased that with this chip technology upgrade, our clients can have more confidence that their benefits will be there when they purchase groceries. This is not the end; we will continue to work and develop new and innovative ways to better protect our clients and their benefits.”
“We are honored to help Alabama DHR lead the way in giving their beneficiaries this critically important tool to protect their accounts and funds,” said Anna Sever, President, Government Solutions at Conduent. “Transitioning to chip technology is a proven fraud-prevention strategy. Chip cards are widely used across the country for other types of accounts, and EBT payments deserve the same level of security.”
SNAP and TANF recipients in Alabama and several other states can also use Conduent’s ConnectEBT mobile app and cardholder portal, which allow beneficiaries to lock their accounts to block all purchases, providing greater control and helping prevent unauthorized transactions.
In addition, with Conduent’s support, Alabama DHR recently implemented a system enhancement that automatically defaults all EBT cards to block out-of-state and online transactions. Cardholders who wish to make these types of purchases can easily unlock their card through the ConnectEBT app or portal.
The technologies are part of Conduent’s VeriSight Anti-Fraud Suite, a set of innovative solutions that help agencies address fraud risks in public benefit programs. The suite includes adaptive fraud detection tools for EBT customer service centers that can identify and block suspicious activity, such as unusual phone numbers or high call volumes.
Conduent is a national leader in government payment disbursements, and it currently supports electronic payments for public programs in 37 states. Conduent also supports U.S. government agencies with end-to-end solutions for healthcare claims processing, eligibility and enrollment, and child support administration.
About Conduent
Conduent delivers digital business solutions and services spanning the commercial, government and transportation spectrum – creating valuable outcomes for its clients and the millions of people who count on them. The Company leverages cloud computing, artificial intelligence, machine learning, automation and advanced analytics to deliver mission-critical solutions. Through a dedicated global team of approximately 51,000 associates, process expertise and advanced technologies, Conduent’s solutions and services digitally transform its clients’ operations to enhance customer experiences, improve performance, increase efficiencies and reduce costs. Conduent adds momentum to its clients’ missions in many ways including disbursing approximately $80 billion in government payments annually, enabling approximately 2.0 billion customer service interactions annually, empowering millions of employees through HR services every year and processing over 14 million tolling transactions every day. Learn more at www.conduent.com.
Conduent is a trademark of Conduent Incorporated in the United States and/or other countries. Other names may be trademarks of their respective owners.
SAN DIEGO, Feb. 13, 2026 (GLOBE NEWSWIRE) — Kratos Defense & Security Solutions, Inc. (NASDAQ: KTOS), a Technology Company in the Defense, National Security and Global Markets, announced today that it will publish financial results for the fourth quarter and fiscal year 2025 after the close of market on Monday, February 23rd. Management will discuss the Company’s operations and financial results in a conference call beginning at 2:00 p.m. Pacific (5:00 p.m. Eastern).
The call will be available at www.kratosdefense.com. Participants may register for the call using this Online Form. Upon registration, all telephone participants will receive the dial-in number along with a unique PIN that can be used to access the call. For those who cannot access the live broadcast, a replay will be available on Kratos’ website.
About Kratos Defense & Security Solutions Kratos Defense & Security Solutions, Inc. (NASDAQ: KTOS) is a technology, products, system and software company addressing the defense, national security, and commercial markets. Kratos makes true internally funded research, development, capital and other investments, to rapidly develop, produce and field solutions that address our customers’ mission critical needs and requirements. At Kratos, affordability is a technology, and we seek to utilize proven, leading edge approaches and technology, not unproven bleeding edge approaches or technology, with Kratos’ approach designed to reduce cost, schedule and risk, enabling us to be first to market with cost effective solutions. We believe that Kratos is known as an innovative disruptive change agent in the industry, a company that is an expert in designing products and systems up front for successful rapid, large quantity, low cost future manufacturing which is a value add competitive differentiator for our large traditional prime system integrator partners and also to our government and commercial customers. Kratos intends to pursue program and contract opportunities as the prime or lead contractor when we believe that our probability of win (PWin) is high and any investment required by Kratos is within our capital resource comfort level. We intend to partner and team with a large, traditional system integrator when our assessment of PWin is greater or required investment is beyond Kratos’ comfort level. Kratos’ primary business areas include virtualized ground systems for satellites and space vehicles including software for command & control (C2) and telemetry, tracking and control (TT&C), jet powered unmanned aerial drone systems, hypersonic vehicles and rocket systems, propulsion systems for drones, missiles, loitering munitions, supersonic systems, space craft and launch systems, C5ISR and microwave electronic products for missile, radar, missile defense, space, satellite, counter UAS, directed energy, communication and other systems, and virtual & augmented reality training systems for the warfighter. For more information, visit www.KratosDefense.com
CHICAGO, Feb. 12, 2026 /PRNewswire/ — Titan International, Inc. announces that Kim Marvin has stepped down from its Board of Directors.
Mr. Marvin stepped down from the Board of Directors of Titan International, Inc. after approximately 24 months of service due to time constraints and other professional commitments. The company currently has no intention of replacing this board seat.
Paul Reitz, President and CEO of Titan International stated “I want to thank Kim for his contributions over the past two years. Kim provided valuable operational continuity following the Carlstar acquisition and Titan benefited from his combination of engineering expertise, financial and transactional experience. We want to wish Kim all the best in his future endeavors.”
About Titan: Titan International, Inc. (NYSE: TWI) is a leading global manufacturer of off-highway wheels, tires, assemblies, and undercarriage products. Headquartered in West Chicago, Illinois, the company globally produces a broad range of products to meet the specifications of original equipment manufacturers (OEMs) and aftermarket customers in the agricultural, earthmoving/construction, and consumer markets. For more information, visit www.titan-intl.com.
Adjusted EBITDA up 15% to $18.5M; Gross Margin expands 210 basis points to 12.8%
Net Income increased to $9.4M, or $0.18 per share, compared to $7.1M, or $0.14 per share, in Q2 FY25
Strengthened balance sheet, ending quarter with $74.1M in working capital
PLANTATION, Fla., Feb. 12, 2026 (GLOBE NEWSWIRE) — Alliance Entertainment Holding Corporation (Nasdaq: AENT), a premier distributor, logistics provider, and omnichannel fulfillment partner to the entertainment and pop culture collectibles industry, supplying more than 340,000 unique SKUs across music, video, video games, licensed merchandise, and exclusive collectibles to over 35,000 retail and e-commerce storefronts, reported its financial and operational results for its fiscal second quarter ended December 31, 2025.
Second Quarter FY 2026 Highlights
Sustained Profitability and Margin Execution: Net income increased year-over-year to approximately $9.4 million, or $0.18 per share, up from $7.1 million, or $0.14 per share in Q2 FY25, reflecting continued execution against the Company’s established profitability baseline. Adjusted EBITDA was approximately $18.5 million, an increase of $2.4 million year-over-year. Adjusted EBITDA margin was approximately 5%, compared to 4.1% in Q2 FY25, a 200 basis point improvement over the margin profile achieved in the trailing 12-months ended September 30, 2025. Gross margin expanded 210 basis points year-over-year to 12.8%, driven by favorable mix and higher-value products. A reconciliation of non-GAAP financial measures to the most comparable GAAP measure is provided at the end of this release.
Launch of Authentication and Digital Product Identity Platform: On December 31, 2025, the Company completed the acquisition of Endstate, establishing Endstate Authentic, a dedicated NFC-enabled authentication and digital product identity platform. The platform expands Alliance’s role beyond physical product distribution by enabling authenticated ownership, provenance, and verified resale across premium physical goods, supporting the full lifecycle of collectible products from initial sale through secondary markets. Designed as a scalable, enterprise-grade platform, Endstate Authentic is intended to support both Alliance’s internal initiatives and third-party brands, licensors, and ecosystem partners, adding a technology-enabled layer that enhances trust, differentiation, and long-term value creation across the collectibles and premium goods market. Subsequent to quarter end, Alliance launched Alliance Authentic™, a premium vinyl collectibles platform that represents the first commercial application of these capabilities within the Company’s portfolio.
Strength in Physical Media: Physical movie revenue increased 33% year-over-year to $114 million, benefiting from sustained demand for premium formats such as 4K Ultra HD and collectible SteelBook editions, as well as the Company’s exclusive distribution partnerships. Alliance was named the exclusive physical media distribution partner for Amazon MGM Studios in North America, effective January 1, 2026, further strengthening its leadership in premium home entertainment and collector-focused releases. Vinyl record sales increased 3% year-over-year, supported by continued consumer demand for collectible and limited-edition releases. Compact disc (CD) sales increased approximately 5% year-over-year, supported by higher unit volumes and the Company’s first full quarter as the exclusive distributor for Virgin Music Group through its AMPED Distribution division.
Collectibles Growth and Portfolio Expansion: Collectibles revenue increased 31% year-over-year, driven by higher average selling prices and a continued shift toward premium, licensed products. Results benefited from expanded sourcing activity, new vendor additions, and the continued integration of the Company’s owned brand, Handmade by Robots™.
Operational Discipline and Infrastructure Investment: Operating income increased year-over-year to $17.3 million, up from $14.8 million in Q2 FY25, reflecting continued operating leverage and disciplined cost management. Total operating expenses rose modestly, driven by targeted investments in technology, personnel, and infrastructure to support exclusive content partnerships and long-term scalability. Distribution and fulfillment costs were 3.3% of net revenue, consistent with 3.2% in Q2 FY25, supported by warehouse automation initiatives and ongoing efficiencies from prior facility consolidation.
Balance Sheet and Liquidity Strength: The Company ended the quarter with working capital of approximately $74.1 million, reflecting disciplined management of inventory and payables. During the quarter, the Company refinanced its asset-based lending agreement with a new $120 million senior secured credit facility from Bank of America, enhancing liquidity and financial flexibility, with availability at quarter end of $35 million.
“Our second quarter results reflect continued execution against the profitability baseline we established last year,” said Jeff Walker, Chief Executive Officer of Alliance Entertainment. “For the six months ended December 31, 2025, earnings per share increased to $0.28, up from $0.15 in the prior-year period, demonstrating the earnings leverage created by our structurally improved margin profile.
“Physical media continues to perform as a collectible category, supported by exclusive partnerships and strong consumer demand for premium formats,” Walker added. “With the launch of Alliance Authentic™, we’re extending that strategy into premium vinyl collectibles by introducing The Ultimate Vinyl Collectible™, enabling fans and collectors to Own a Piece of Vinyl History™ through authentic, certified, and individually numbered releases sourced directly from rights holders. This initiative builds on our strengths in physical media and reinforces our focus on high-value, enthusiast-driven products. With a structurally stronger margin profile and a growing pipeline of exclusive content, we believe Alliance is well positioned to deliver durable profitability and long-term value for our shareholders.”
Amanda Gnecco, Chief Financial Officer of Alliance Entertainment, said, “Net income in the second quarter increased 33% year-over-year to $9.4 million, and adjusted EBITDA margin improved 92 basis points year-over-year to 5.0%, reflecting the durability of our cost structure and the benefits of our improving product mix.
“During the quarter, we strengthened our balance sheet by refinancing our credit facility with Bank of America, reducing borrowing costs by up to 250 basis points and extending the maturity to five years. We ended the quarter with just over $74 million in working capital and enhanced liquidity, providing greater financial flexibility to support premium inventory, exclusive partnerships, and strategic initiatives while maintaining disciplined capital management,” continued Gnecco.
“As we look ahead, we’re building on a much stronger foundation,” Walker continued. “The acquisition of Endstate and the launch of Endstate Authentic mark an important step in expanding Alliance beyond distribution into authenticated collectibles, digital product identity, and recurring platform-driven revenue. This technology allows us to extend the value of physical products across their entire lifecycle-from initial sale through authenticated resale-while strengthening trust, provenance, and margins across our ecosystem. With the launch of Alliance Authentic™, we are also creating new opportunities in the collectible vinyl market by applying authentication, scarcity, and provenance to products we already source and distribute at scale.
“Separately, our new exclusive partnership with Amazon MGM Studios strengthens our leadership in premium physical home entertainment,” Walker added. “By combining our scale, operational execution, and exclusive studio relationships, we continue to elevate physical movies as collectible formats for fans and enthusiasts. Together, these initiatives reflect a disciplined approach to growth that leverages our scale, exclusivity, and financial flexibility to create long-term shareholder value.”
Second Quarter FY 2026 Financial Results
Net revenues for the fiscal second quarter ended December 31, 2025, were $369 million, compared to $394 million in the same period of fiscal 2025.
Gross profit for the fiscal second quarter ended December 31, 2025, was $47.1 million, compared to $42.3 million in the same period of fiscal 2025.
Gross margin for the fiscal second quarter ended December 31, 2025, was 12.8%, up 210 basis points from 10.7% in the same period of fiscal 2025.
Net income for the fiscal second quarter ended December 31, 2025, was $9.4 million, or $0.18 per diluted share, compared to net income of $7.1 million, or $0.14 per diluted share for the same period of fiscal 2025.
Adjusted EBITDA for the fiscal second quarter ended December 31, 2025, was $18.5 million, compared to Adjusted EBITDA of $16.1 million for the same period of fiscal 2025.
Six-Months FY 2026 Financial Results
Net revenues for the six months ended December 31, 2025, were $623 million, compared to $623 million in the same period of fiscal 2025.
Gross profit for the six months ended December 31, 2025, was $84.3 million, compared to $67.8 million in the same period of fiscal 2025.
Gross margin for the six months ended December 31, 2025, was 13.5%, up 260 basis points from 10.9% in the same period of fiscal 2025.
Net income for the six months ended December 31, 2025, was $14.3 million, or $0.28 per diluted share, compared to net income of $7.5 million, or $0.15 per diluted share for the same period of fiscal 2025.
Adjusted EBITDA for the six months ended December 31, 2025, was $30.7 million, compared to Adjusted EBITDA of $19.5 million for the same period of fiscal 2025.
Conference Call
Alliance Entertainment Chief Executive Officer Jeff Walker, Chief Financial Officer Amanda Gnecco, and Executive Chairman Bruce Ogilvie will host the conference call, which will be followed by a question-and-answer session. A presentation will accompany the call and can be viewed during the webcast or accessed via the investor relations section of the Company’s website here.
To access the call, please use the following information:
Date:
Thursday, February 12, 2026
Time:
4:30 p.m. Eastern Time, 1:30 p.m. Pacific Time
Toll-free dial-in number:
1-877-407-0784
International dial-in number:
1-201-689-8560
Conference ID:
13758224
Please call the conference telephone number 5-10 minutes prior to the start time. An operator will register your name and organization. If you have any difficulty connecting with the conference call, please contact RedChip Companies at 1-407-644-4256.
A telephone replay of the call will be available approximately three hours after the call concludes and can be accessed through March 12, 2026, using the following information:
Toll-free replay number:
1-844-512-2921
International replay number:
1-412-317-6671
Replay ID:
13758224
About Alliance Entertainment
Alliance Entertainment (NASDAQ: AENT) is a premier distributor and fulfillment partner for the entertainment and pop culture collectibles industry. With more than 340,000 unique in-stock SKUs – including over 57,300 exclusive titles across compact discs, vinyl LPs, DVDs, Blu-rays, and video games – Alliance offers the largest selection of physical media in the market. Our vast catalog also includes licensed merchandise, toys, retro gaming products, and collectibles, serving over 35,000 retail locations and powering e-commerce fulfillment for leading retailers. Alliance also owns and operates proprietary collectibles brands, including Handmade by Robots™, a stylized vinyl figure line featuring licensed characters from leading entertainment franchises, and Alliance Authentic™, a premium platform for authentic, certified, and individually numbered entertainment collectibles. In addition, Alliance operates Endstate Authentic, a dedicated NFC-enabled authentication and digital product identity platform supporting authenticated collectibles, resale, and brand protection. Leveraging decades of operational expertise, exclusive sourcing relationships, and a capital-light, scalable infrastructure, Alliance connects fans and collectors to the products, franchises, and experiences they value across formats and generations. For more information, visit www.aent.com.
Forward Looking Statements
Certain statements included in this Press Release that are not historical facts are forward-looking statements for purposes of the safe harbor provisions under the United States Private Securities Litigation Reform Act of 1995. Forward-looking statements generally are accompanied by words such as “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “expect,” “should,” “would,” “plan,” “predict,” “potential,” “seem,” “seek,” “future,” “outlook,” and similar expressions that predict or indicate future events or trends or that are not statements of historical matters. These forward-looking statements include, but are not limited to, statements regarding estimates and forecasts of other financial and performance metrics and projections of market opportunity. These statements are based on various assumptions, whether identified in this Press Release, and on the current expectations of Alliance’s management and are not predictions of actual performance. These forward-looking statements are provided for illustrative purposes only and are not intended to serve as and must not be relied on by an investor as, a guarantee, an assurance, a prediction, or a definitive statement of fact or probability. Actual events and circumstances are difficult or impossible to predict and will differ from assumptions. Many actual events and circumstances are beyond the control of Alliance. These forward-looking statements are subject to a number of risks and uncertainties, including risks relating to the anticipated growth rates and market opportunities; changes in applicable laws or regulations; the ability of Alliance to execute its business model, including market acceptance of its systems and related services; Alliance’s reliance on a concentration of suppliers for its products and services; increases in Alliance’s costs, disruption of supply, or shortage of products and materials; Alliance’s dependence on a concentration of customers, and failure to add new customers or expand sales to Alliance’s existing customers; increased Alliance inventory and risk of obsolescence; Alliance’s significant amount of indebtedness; our ability to refinance our existing indebtedness; our ability to continue as a going concern absent access to sources of liquidity; risks that a breach of the revolving credit facility could result in the lender declaring a default and that the full outstanding amount under the revolving credit facility could be immediately due in full, which would have severe adverse consequences for the Company; known or future litigation and regulatory enforcement risks, including the diversion of time and attention and the additional costs and demands on Alliance’s resources; Alliance’s business being adversely affected by increased inflation, uncertainty regarding tariffs, higher interest rates and other adverse economic, business, and/or competitive factors; geopolitical risk and changes in applicable laws or regulations; as well as our financial condition and results of operations; substantial regulations, which are evolving, and unfavorable changes or failure by Alliance to comply with these regulations; product liability claims, which could harm Alliance’s financial condition and liquidity if Alliance is not able to successfully defend or insure against such claims; availability of additional capital to support business growth; and the inability of Alliance to develop and maintain effective internal controls.
For investor inquiries, please contact:
Dave Gentry RedChip Companies, Inc. 1-800-REDCHIP (733-2447) 1-407-644-4256 [email protected]
HOUSTON, Feb. 12, 2026 /PRNewswire/ — Direct Digital Holdings, Inc. (Nasdaq: DRCT) (“Direct Digital Holdings” or the “Company”), a leading advertising and marketing technology platform operating through its companies Colossus Media, LLC (“Colossus SSP”) and Orange 142, LLC (“Orange 142”), today announced that the Company has received formal notice from The Nasdaq Stock Market LLC (“Nasdaq”) confirming that the Company has regained compliance with Nasdaq Listing Rule 5550(a)(2), which requires a minimum bid price of $1.00 per share, and otherwise satisfies all applicable criteria for continued listing on The Nasdaq Capital Market.
Mark Walker, CEO of Direct Digital Holdings, commented, “Evidencing full compliance with the Nasdaq listing criteria represents an important step on our path forward and the continued execution on our strategic goals.”
The Company’s common stock will continue to trade on Nasdaq under the ticker symbol “DRCT.”
This press release contains forward-looking statements within the meaning of federal securities laws that are subject to certain risks, trends and uncertainties. We use words such as “could,” “would,” “may,” “might,” “will,” “expect,” “likely,” “believe,” “continue,” “anticipate,” “estimate,” “intend,” “plan,” “project” and other similar expressions to identify forward-looking statements, but not all forward-looking statements include these words. All of our forward-looking statements involve estimates and uncertainties that could cause actual results to differ materially from those expressed in or implied by the forward-looking statements. Accordingly, any such statements are qualified in their entirety by reference to the information described under the caption “Risk Factors” and elsewhere in our most recent Annual Report on Form 10-K for the fiscal year ended December 31, 2024 (the “Form 10-K”) and subsequent periodic and or current reports filed with the Securities and Exchange Commission (the “SEC”).
The forward-looking statements contained in this press release are based on assumptions that we have made in light of our industry experience and our perceptions of historical trends, current conditions, expected future developments and other factors we believe are appropriate under the circumstances. As you read and consider this press release, you should understand that these statements are not guarantees of performance or results. They involve risks, uncertainties (many of which are beyond our control) and assumptions.
Although we believe that these forward-looking statements are based on reasonable assumptions, you should be aware that many factors could affect our actual operating and financial performance and cause our performance to differ materially from the performance expressed in or implied by the forward-looking statements. We believe these factors include, but are not limited to, the following: the restrictions and covenants imposed upon us by our credit facilities; the substantial doubt about our ability to continue as a going concern, which may hinder our ability to obtain future financing; our ability to secure additional financing to meet our capital needs; our ability to maintain compliance with applicable listing standards of the Nasdaq Capital Market; any significant fluctuations caused by our high customer concentration; risks related to non-payment by our clients; reputational and other harms caused by our failure to detect advertising fraud; operational and performance issues with our platform, whether real or perceived, including a failure to respond to technological changes or to upgrade our technology systems; restrictions on the use of third-party “cookies,” mobile device IDs or other tracking technologies, which could diminish our platform’s effectiveness; unfavorable publicity and negative public perception about our industry, particularly concerns regarding data privacy and security relating to our industry’s technology and practices, and any perceived failure to comply with laws and industry self-regulation; our failure to manage our growth effectively; the difficulty in identifying and integrating any future acquisitions or strategic investments; any changes or developments in legislative, judicial, regulatory or cultural environments related to information collection, use and processing; challenges related to our buy-side clients that are destination marketing organizations and that operate as public/private partnerships; any strain on our resources or diversion of our management’s attention as a result of being a public company; the intense competition of the digital advertising industry and our ability to effectively compete against current and future competitors; any significant inadvertent disclosure or breach of confidential and/or personal information we hold, or of the security of our or our customers’, suppliers’ or other partners’ computer systems; as a holding company, we depend on distributions from Direct Digital Holdings, LLC (“DDH LLC”) to pay our taxes, expenses (including payments under the Tax Receivable Agreement) and any amount of any dividends we may pay to the holders of our common stock; any failure by us to maintain or implement effective internal controls or to detect fraud; and other factors and assumptions discussed in our Form 10-K and subsequent periodic and current reports we may file with the SEC.
Should one or more of these risks or uncertainties materialize or should any of these assumptions prove to be incorrect, our actual operating and financial performance may vary in material respects from the performance projected in these forward-looking statements. Further, any forward-looking statement speaks only as of the date on which it is made, and except as required by law, we undertake no obligation to update any forward-looking statement contained in this press release to reflect events or circumstances after the date on which it is made or to reflect the occurrence of anticipated or unanticipated events or circumstances, and we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995. New factors that could cause our business not to develop as we expect emerge from time to time, and it is not possible for us to predict all of them. Further, we cannot assess the impact of each currently known or new factor on our results of operations or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
About Direct Digital Holdings
Direct Digital Holdings (Nasdaq: DRCT) combines cutting-edge sell-side and buy-side advertising solutions, providing data-driven digital media strategies that enhance reach and performance for brands, agencies, and publishers of all sizes. Our sell-side platform, Colossus SSP, offers curated access to premium, growth-oriented media properties throughout the digital ecosystem. On the buy-side, Orange 142 delivers customized, audience-focused digital marketing and advertising solutions that enable mid-market and enterprise companies to achieve measurable results across a range of platforms, including programmatic, search, social, CTV, and influencer marketing. With extensive expertise in high-growth sectors such as Energy, Healthcare, Travel & Tourism, and Financial Services, our teams deliver performance strategies that connect brands with their ideal audiences.
At Direct Digital Holdings, we prioritize personal relationships by humanizing technology, ensuring each client receives dedicated support and tailored digital marketing solutions regardless of company size. This empowers everyone to thrive by generating billions of monthly impressions across display, CTV, in-app, and emerging media channels through advanced targeting, comprehensive data insights, and cross-platform activation. DDH is “Digital advertising built for everyone.”
Earnings Release: Tuesday, February 17, 2026, Before Market Open in New York Conference Call and Webcast: Tuesday, February 17, 2026, at 10:00 a.m. Eastern Time
GLYFADA, Greece, Feb. 12, 2026 (GLOBE NEWSWIRE) — Seanergy Maritime Holdings Corp. (the “Company” or “Seanergy”) (NASDAQ: SHIP) announced today that it will release its financial results for the fourth quarter and year ended December 31, 2025, prior to the open of the market in New York on Tuesday, February 17, 2026.
Seanergy’s senior management will conduct a conference call and simultaneous Internet webcast to review these results on Tuesday, February 17, 2026, at 10:00 a.m. Eastern Time.
Audio Webcast and Earnings Presentation: There will be a live, and then archived, webcast of the conference call and accompanying slides available through the Company’s website. To access the slides and listen to the archived audio file, visit our website, following the Webcast & Presentations section under our Investor Relations page. Participants to the live webcast should register on Seanergy’s website approximately 10 minutes prior to the start of the webcast, by following this link.
Conference Call Details: Participants have the option to register for the call using the following link. You can use any number from the list or add your phone number and let the system call you right away.
About Seanergy Maritime Holdings Corp. Seanergy Maritime Holdings Corp. is a prominent pure-play Capesize shipping company publicly listed in the U.S. Seanergy provides marine dry bulk transportation services through a modern fleet of Capesize vessels. The Company’s operating fleet consists of 20 vessels (2 Newcastlemax and 18 Capesize) with an average age of approximately 14.6 years and an aggregate cargo carrying capacity of approximately 3,633,861 dwt.
The Company is incorporated in the Republic of the Marshall Islands and has executive offices in Glyfada, Greece. The Company’s common shares trade on the Nasdaq Capital Market under the symbol “SHIP”.
Forward-Looking Statements This press release contains forward-looking statements (as defined in Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended) concerning future events, including with respect to declaration of dividends, market trends and shareholder returns. Words such as “may”, “should”, “expects”, “intends”, “plans”, “believes”, “anticipates”, “hopes”, “estimates” and variations of such words and similar expressions are intended to identify forward-looking statements. These statements involve known and unknown risks and are based upon a number of assumptions and estimates, which are inherently subject to significant uncertainties and contingencies, many of which are beyond the control of the Company. Actual results differ materially from those expressed or implied by such forward-looking statements. Factors that could cause actual results to differ materially include, but are not limited to, the Company’s operating or financial results; the Company’s liquidity, including its ability to service its indebtedness; competitive factors in the market in which the Company operates; shipping industry trends, including charter rates, vessel values and factors affecting vessel supply and demand; future, pending or recent acquisitions and dispositions, business strategy, impacts of litigation, areas of possible expansion or contraction, and expected capital spending or operating expenses; risks associated with operations outside the United States; broader market impacts arising from trade disputes or war (or threatened war) or international hostilities, such as between Israel and Hamas or Iran, China and Taiwan and between Russia and Ukraine; risks associated with the length and severity of pandemics, including their effects on demand for dry bulk products and the transportation thereof; and other factors listed from time to time in the Company’s filings with the SEC, including its most recent annual report on Form 20-F. The Company’s filings can be obtained free of charge on the SEC’s website at www.sec.gov. Except to the extent required by law, the Company expressly disclaims any obligations or undertaking to release publicly any updates or revisions to any forward-looking statements contained herein to reflect any change in the Company’s expectations with respect thereto or any change in events, conditions or circumstances on which any statement is based.
For further information please contact: Seanergy Investor Relations Tel: +30 213 0181 522 E-mail: [email protected]
Capital Link, Inc. Paul Lampoutis 230 Park Avenue Suite 1540 New York, NY 10169 Tel: (212) 661-7566 Email: [email protected]
GROSSE POINTE FARMS, Mich., Feb. 12, 2026 (GLOBE NEWSWIRE) — Saga Communications, Inc. (Nasdaq – SGA) (the “Company”, “Saga” or “our”) today announced that its Board of Directors (“Board”) declared a quarterly cash dividend of $0.25 per share. The dividend will be paid on March 20, 2026, to shareholders of record on February 26, 2026. The aggregate amount of the payment to be made in connection with the quarterly dividend will be approximately $1.6 million. The quarterly dividend will be funded by cash on the Company’s balance sheet. Including this dividend, the Company will have paid over $143 million in dividends to shareholders since the first special dividend was paid in 2012.
The Company currently intends to declare regular quarterly cash dividends in the future. Further, as part of its overall capital allocation plan for fiscal year 2026 the Company may also implement stock buybacks and declare special dividends in future periods. The declaration and payment of any future dividend, whether fixed, special, or based on the variable policy, or the implementation of any stock buyback program will remain at the full discretion of the Board and will depend on the Company’s financial results, cash requirements, future expectations, and other pertinent factors.
Saga is a media company whose business provides radio, digital, e-commerce, local on-line news and non-traditional revenue initiatives. Saga operates in 28 markets and provides services to national, regional and local advertisers to meet their growing advertising needs. For additional information, contact us at (313) 886-7070 or visit our website at www.sagacom.com.
This press release contains certain forward-looking statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 that are based upon current expectations and involve certain risks and uncertainties. Words such as “will,” “may,” “believes,” “intends,” “expects,” “anticipates,” “guidance,” and similar expressions are intended to identify forward-looking statements. The material risks facing our business are described in the reports Saga periodically files with the U.S. Securities and Exchange Commission, including, in particular, Item 1A of our Annual Report on Form 10-K. Readers should note that forward-looking statements may be impacted by several factors, including global, national, and local economic changes and changes in the radio broadcast industry in general as well as Saga’s actual performance. Actual results may vary materially from those described herein and Saga undertakes no obligation to update any information contained herein that constitutes a forward-looking statement.