DALLAS–(BUSINESS WIRE)–Jul. 8, 2026– Resources Connection, Inc. (Nasdaq: RGP) (the “Company,” “we,” and “our”), a global consulting firm, will announce results of operations for its fourth quarter and full fiscal year ended May 30, 2026 after the close of market on July 22, 2026.
This release will be followed by a conference call at 5:00 p.m. ET, July 22, 2026. A live webcast of the call will be available on the “Investor Relations” Events section of the Company’s website. To access the call by phone, please go to this link (registration link), and you will be provided with dial in details. To avoid delays, we encourage participants to dial into the conference call fifteen minutes ahead of the scheduled start time. A replay of the webcast will also be available for a limited time by visiting the RGP Investor Events section of the Company’s website.
ABOUT RGP
RGP (Nasdaq: RGP) has been redefining professional services for 30 years by closing the gap between advice and execution. RGP combines the flexibility of on-demand talent, the rigor of consulting, and the accountability of managed services for faster impact, smarter investment, and lower risk. The firm partners with CFOs and other C-suite leaders across finance, digital transformation, data, and cloud — connecting advisory to execution at global scale.
Based in Dallas, Texas, with offices worldwide, RGP annually engages with more than 1,500 clients around the world from 40 physical practice offices and multiple virtual offices. As of January 2026, RGP is proud to have served 90 percent of the Fortune 100 and has been recognized by U.S. News & World Report (2025–2026 Best Companies to Work For) and Forbes (America’s Best Midsize Employers 2026, America’s Best Management Consulting Firms 2025, World’s Best Management Consulting Firms 2025).
The Company is listed on the Nasdaq Global Select Market, the exchange’s highest tier by listing standards. To learn more about RGP, visit: http://www.rgp.com.
All references to dollar amounts are references to U.S. Dollars, unless otherwise stated
Toronto, Ontario–(Newsfile Corp. – July 21, 2026) – Kuya Silver Corporation (CSE: KUYA) (OTCQB: KUYAF) (FSE: 6MR1) (the “Company” or “Kuya Silver“) is pleased to report record quarterly production and provide an operational update for the second quarter of 2026 at the Bethania silver project, which again delivered record daily and quarterly production rates as the ramp-up continued to show significant progress during the quarter. Although the short-term progress at Bethania is encouraging, preparations are underway for upward step change in production later this year as additional underground development is completed.
Operational Highlights
5,097 metric tonnes of mineralized material mined at Bethania, a 66% increase quarter over quarter.
Record 23,912 oz silver (30,559 silver equivalent) processed during the quarter.
Record monthly production in June of 13,273 silver equivalent ounces as grades improved toward the end of the quarter.
Continued strong underground development with record 437 meters advanced while drilling and blasting 1,535 metric tonnes of development material to support the expansion of underground mining operations.
Silver production was 87% of the quarterly revenue from Bethania in Q2 with an average selling price of $72/oz.
The Company continued to develop partnerships with local contractors to accelerate our exploration and mine development objectives.
Kuya Silver Delivers Steady Progress at Bethania in Q2
Production of mineralized material at the Bethania Project totalled 5,097 metric tonnes, another quarterly record and a 66% improvement over Q1 2026 as production continues to steadily ramp up. Development activities achieved total of 437 metres of underground advancement and 1,535 tonnes of development material in the quarter. Kuya Silver achieved a record daily production of 124 tonnes and a record monthly production of 2,040 tonnes (68 tpd) in Q2, as well as a monthly record for silver production (13,273 oz Ag eq.) demonstrating that the Company’s methodical ramp-up process is generating positive results.
Silver recoveries averaged 79.7% during Q2 2026, directly reflecting the lower-grade development material and stope scheduling early in the quarter, which resulted in an average grade of 5.9 oz/t silver (8.8 oz/t or 274 g/t Ag equivalent). Mine sequencing optimizations implemented by the Kuya Silver team began delivering positive results mid-quarter. By June, silver grades increased to 6.66 oz/t exceeding management’s expectations, and silver recoveries improved to approximately 82%. Kuya Silver has previously achieved silver recoveries exceeding 90% when processing higher-grade batches, the Company expects recoveries to continue improving toward these levels as the mine reaches steady-state production. Furthermore, Kuya Silver has launched a metallurgical testing campaign to further optimize recoveries.
In addition, Kuya Silver approved the installation of a dual-car hoisting winch system at Bethania, expected to be commissioned in October 2026. This infrastructure upgrade is expected to improve efficiency of the mine’s ore extraction by enabling simultaneous haulage of two ore cars. This improvement will support the ongoing production ramp-up and provide flexibility and redundancy to materials handling as the Company advances the underground ramp project.
David Stein, Kuya Silver’s President and CEO, stated, “The ramp up strategy to produce at the Bethania mine while taking on ambitious mine development to expand production to our Phase 1 target of 350 tpd has allowed the Company to generate significant revenue and reduce our burn rate. The revenue from mining operations on top of the Company’s already strong balance sheet puts Kuya Silver in an excellent position, for the first time in our history, to deliver on its near-term production targets while at the same time expanding our exploration efforts to delineate more silver at the Bethania mine and the six other silver veins systems we control in the Bethania district.”
Table 1: Production highlights from the Bethania silver mine
(1)Information has been revised from amounts previously disclosed in the Company’s press release dated April 22, 2026. The revisions relate primarily to the correction of previously reported production metrics.
(2)prices for silver equivalent calculations use period ending spot prices and are as follows: June 30, 2026 $61/oz, lead $1,877/tonne, zinc$3,552/tonne Mar. 31 2026 silver $74/oz, lead $1,909/tonne, zinc$3,230/tonne June 30, 2025 $36/oz, lead $2,205/tonne, zinc$2,764/tonne Mar. 31 2026 silver $74/oz, lead $1,909/tonne Mar. 31, 2025 period; silver $34/oz, gold $3122.80/oz, lead $2,002/tonne, zinc $2,829/tonne.
(3)includes only payable recovery i.e. lead in the silver- lead concentrate and zinc in the zinc concentrate and silver in both concentrates.
(4)may include provisional settlements at the end of the period, net of treatment and refining costs.
Developing Partnerships with Local Mining and Exploration Contractors
As part of its strategy to sustain production growth and expand exploration efforts at the Bethania project, Kuya Silver is in the process of onboarding up to three specialized contractors to support development, drilling and operations at the project. The underground drilling contract to operate three drill rigs at Bethania has been formally awarded to Safasermin S.A.C., an experienced Peruvian drilling and exploration contractor, pending the finalization of contract details. All three rigs are initially planned to operate underground at the Bethania mine; however, surface drilling is expected to be added and become a significant component of the drilling program during the second half of 2026.
Following a competitive bidding process, comprehensive site visits and final negotiations, Kuya Silver has issued a formal Letter of Award to Minera Tauro S.A.C. to perform additional underground development at the Bethania mine. The contractor has officially confirmed its acceptance. Pending final execution of the contract, Minera Tauro is expected to mobilize in the coming weeks to accelerate underground development. This partnership will allow Kuya Silver’s in-house operations staff to focus on achieving greater production levels as the ramp-up continues.
Kuya Silver has also commenced portal development for the new internal ramp project and advanced a rigorous, competitive bidding process to shift the majority of the ramp development to a specialized mining contractor. Following comprehensive site visits and detailed evaluations of the project’s technical data and Terms of Reference, Kuya Silver has finalized the receipt of complete technical and economic proposals from four highly respected mining contractors. The Company is currently evaluating the final submissions and expects to formally award the contract and mobilize the selected contractor shortly.
In parallel, Kuya Silver has completed a geomechanical assessment and ground-support design study for the ramp project, characterizing rock-mass conditions and establishing the optimal reinforcement system for each section of the planned corridor. This engineering work enables the Company to move directly from contractor award to mobilization without delay. The preparatory work is expected to provide greater confidence in both the execution timeline and the cost profile of this critical infrastructure investment.
Christian Aramayo, Kuya Silver’s Chief Operating Officer, remarked, “At Bethania, our development decisions are driven by geology, not short-term price volatility. We are partnering with elite contractors and expanding our team to optimize development ahead of completing our Phase 1 ramp-up. We are committing to disciplined, high-return infrastructure, such as advancing our new internal ramp project, to unlock the deeper, and higher-grade vein structures within the Bethania system. Our strong balance sheet allows us to build our infrastructure the right way, ensuring we develop a safe and flexible operation, capable of maximizing the margin of every ounce we mine.”
Camila Plant Update
Kuya Silver continues to process its mineralized material at the Camila Plant. The Company announced an LOI on January 27, 2026 (see press release) and plans to make a separate announcement.
Support for Local Districts and Earthquake Relief Efforts
While Kuya Silver’s Bethania mine and infrastructure reported no measurable impact from the July 19th 5.5 magnitude earthquake in the Junin region, we recognize the severe toll this seismic event has taken on the surrounding towns and districts, particularly in the district of Chongos Bajo and the highly affected area of Chupuro. Kuya Silver extends its deepest sympathies to the people of Junin and is actively working to support recovery efforts.
As part of our commitment to our local workforce and neighboring populations, Kuya Silver is providing direct assistance to our employees who have family members in the affected areas. Furthermore, the Company is making direct donations to support the local emergency relief efforts.
For our partners, contractors, and stakeholders who wish to join these relief efforts, local institutions have established official donation reception centers to support the victims. Donations of drinking water, non-perishable food, warm clothing, and essential medicines are currently being received at the following collection point: Municipalidad Distrital de Chongos Bajo: Main municipal offices.
Quality Assurance and Quality Control
Quality assurance and quality control include two sampling procedures. Underground vein material from stopes are sampled to confirm vein grades and to reconcile against the mine model; and sampling of freshly mined material in stockpiles to determine dilution and the head grade that is sent to the processing plant.
Underground vein sampling was conducted systematically every 4 meters along the galleries. This involved excavating a narrow and continuous channel either parallel to the vein or perpendicular to its orientation. The entire volume of material excavated from the channel was collected as a sample.
Freshly mined material in the stockpiles and concentrate stockpiles were sampled using trenching, a method involving the excavation of narrow trenches perpendicular to the major axis of the pile. Trenches were systematically dug at regular intervals across all depths of the pile. The location of each trench was referenced to a topographic control point and recorded in the sampling log.
All material was carefully collected on plastic sheets, then pulverized at the mine site. The pulverized material was quartered, and one quarter was labeled and secured in vinyl sample bags. The samples were then transported to Dmtri I. Mendelejeff laboratory in Huancayo for processing using fire assay followed by atomic absorption spectroscopy (AAS).
All concentrate assay results are cross-checked against independent analyses conducted by the buyer. Furthermore, sample security protocols include sealed trucks for transporting run-of-mine (ROM) material and concentrate trucks with tamper-proof devices with safety seals, and a documented custody chain overseen by the mine superintendent (Bethania).
National Instrument 43-101 Disclosure
The technical content of this news release has been reviewed and approved by Mr. Kevin J. O’Connell, P.E., Independent Technical Advisor to of Kuya Silver and a Qualified Person as defined by National Instrument 43-101.
About Kuya Silver Corporation
Kuya Silver is a Canadian‐based, growth-oriented mining company with a focus on silver. Kuya Silver operates the Bethania silver mine in Peru, while developing district-scale silver projects in mining-friendly jurisdictions including Peru and Canada.
This news release contains statements that constitute “forward-looking information,” including statements regarding the plans, intentions, beliefs, and current expectations of the Company, its directors, or its officers with respect to the future business activities of the Company. The words “may,” “would,” “could,” “will,” “intend,” “plan,” “anticipate,” “believe,” “estimate,” “expect,” “must,” “next,” “propose,” “new,” “potential,” “prospective,” “target,” “future,” “verge,” “favorable,” “implications,” and “ongoing,” and similar expressions, as they relate to the Company or its management, are intended to identify such forward-looking information. Investors are cautioned that statements including forward-looking information are not guarantees of future business activities and involve risks and uncertainties, and that the Company’s future business activities may differ materially from those described in the forward-looking information as a result of various factors, including but not limited to fluctuations in market prices, successes of the operations of the Company, continued availability of capital and financing, and general economic, market, and business conditions. There can be no assurances that such forward-looking information will prove accurate, and therefore, readers are advised to rely on their own evaluation of the risks and uncertainties. The Company does not assume any obligation to update any forward-looking information except as required under the applicable securities laws.
Neither the Canadian Securities Exchange nor the Investment Industry Regulatory Organization of Canada accepts responsibility for the adequacy or accuracy of this release.
ASPR Report Reinforces National Priorities for Domestic Manufacturing, Platform Technologies, Strategic National Stockpile Modernization, and Pandemic Preparedness
ATLANTA, GA – July 21, 2026 – GeoVax Labs, Inc. (Nasdaq: GOVX), a clinical-stage biotechnology company developing next-generation vaccines and immunotherapies, today highlighted the strong alignment between its GEO-MVA vaccine platform strategy and the priorities outlined in the U.S. Department of Health and Human Services Administration for Strategic Preparedness and Response’s (ASPR) newly released Public Health Emergency Medical Countermeasure Enterprise (PHEMCE) Multiyear Budget: Fiscal Years 2025–2029.
The report provides the federal government’s five-year roadmap for investments in medical countermeasures, advanced manufacturing, Strategic National Stockpile (SNS) modernization, and preparedness against emerging infectious disease and biological threats.
According to the report, approximately 65% of future medical countermeasure investment is expected to focus on threat-agnostic and multi-threat technologies, while emphasizing domestic manufacturing capability, scalable vaccine platforms, and sustained preparedness infrastructure. The report also projects a total five-year medical countermeasure investment need of approximately $66.9 billion, underscoring the growing strategic importance of resilient domestic biodefense capabilities.
“The release of ASPR’s five-year preparedness strategy reinforces many of the strategic priorities that have guided our GEO-MVA program,” said David A. Dodd, Chairman and Chief Executive Officer of GeoVax. “The report recognizes that future preparedness requires more than individual vaccines – it requires flexible platform technologies, domestic manufacturing capability, resilient supply chains, and sustainable medical countermeasure infrastructure. GEO-MVA has been developed with these national priorities in mind.”
ASPR Priorities Closely Align with the GEO-MVA Strategy
GeoVax believes several priorities identified in the ASPR strategy directly support the long-term strategic rationale underlying the GEO-MVA program.
Domestic Manufacturing
The ASPR report identifies sustaining and expanding domestic commercial manufacturing capability as one of the nation’s principal preparedness challenges.
GeoVax’s GEO-MVA program is designed to establish a U.S.-based source of Modified Vaccinia Ankara (MVA) vaccine manufacturing, supporting domestic supply-chain resilience while reducing dependence on limited global manufacturing capacity.
Platform Technologies
The report emphasizes investments in threat-agnostic and multi-threat technologies capable of responding rapidly to future public health emergencies.
The MVA vector represents a well-established vaccine platform with demonstrated applicability across multiple infectious disease targets, providing flexibility beyond any single indication.
Strategic National Stockpile
The ASPR strategy calls for continued modernization of the Strategic National Stockpile while addressing lifecycle management, replenishment, and transition of new medical countermeasures into long-term preparedness programs.
GeoVax believes the GEO-MVA program aligns with these objectives by supporting future U.S. preparedness capacity for orthopoxvirus threats.
Emerging Infectious Diseases
The report identifies emerging infectious diseases as among the nation’s highest preparedness priorities while emphasizing investments that enable rapid response to evolving threats.
GeoVax’s clinical development strategy for GEO-MVA is intended to support preparedness against mpox and smallpox while leveraging the broader capabilities of the MVA platform.
Independent Validation of Long-Term Preparedness Trends
The ASPR report reflects a continuing evolution in federal preparedness strategy – from investing primarily in individual products toward investing in strategic capabilities, including manufacturing infrastructure, platform technologies, and sustainable preparedness systems.
GeoVax believes these evolving priorities reinforce the long-term strategic value of domestic vaccine manufacturing capabilities that can support future public health emergency response.
“Preparedness is increasingly being viewed as a strategic national capability rather than simply a collection of products,” added Mr. Dodd. “We believe this evolution supports the importance of maintaining flexible vaccine platforms and domestic manufacturing infrastructure capable of responding to future biological threats.”
About GEO-MVA
GEO-MVA is GeoVax’s Modified Vaccinia Ankara (MVA) vaccine candidate being developed to support protection against mpox and smallpox. The program is advancing through an immunobridging regulatory strategy following positive Scientific Advice from the European Medicines Agency (EMA) and is intended to provide an additional source of MVA vaccine manufacturing capacity supporting future public health preparedness initiatives.
About GeoVax
GeoVax Labs, Inc. is a clinical-stage biotechnology company focused on the development of vaccines and immunotherapies addressing high-consequence infectious diseases and solid tumor cancers. GeoVax’s priority program is GEO-MVA, a Modified Vaccinia Ankara (MVA)–based vaccine targeting mpox and smallpox. The program is advancing under an expedited regulatory pathway, with plans to initiate a pivotal Phase 3 clinical trial in the second half of 2026, to address critical global needs for expanded orthopoxvirus vaccine supply and biodefense preparedness. In oncology, GeoVax is developing Gedeptin®, a gene-directed enzyme prodrug therapy (GDEPT) designed to enhance immune checkpoint inhibitor activity. Gedeptin has completed a multicenter Phase 1/2 clinical trial in advanced head and neck cancer and is being advanced into combination strategies, including planned neoadjuvant and first-line settings. GeoVax maintains a global intellectual property portfolio supporting its infectious disease and oncology programs and continues to evaluate strategic partnerships and funding opportunities aligned with its development priorities. For more information, visit www.geovax.com.
Forward-Looking Statements
This release contains forward-looking statements regarding GeoVax’s business plans. The words “believe,” “look forward to,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “could,” “target,” “potential,” “is likely,” “will,” “expect” and similar expressions, as they relate to us, are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. Actual results may differ materially from those included in these statements due to a variety of factors, including whether: GeoVax is able to obtain acceptable results from ongoing or future clinical trials of its investigational products, GeoVax’s immuno-oncology products and preventative vaccines can provoke the desired responses, and those products or vaccines can be used effectively, GeoVax’s viral vector technology adequately amplifies immune responses to cancer antigens, GeoVax can develop and manufacture its immuno-oncology products and preventative vaccines with the desired characteristics in a timely manner, GeoVax’s immuno-oncology products and preventative vaccines will be safe for human use, GeoVax’s vaccines will effectively prevent targeted infections in humans, GeoVax’s immuno-oncology products and preventative vaccines will receive regulatory approvals necessary to be licensed and marketed, GeoVax raises required capital to complete development, there is development of competitive products that may be more effective or easier to use than GeoVax’s products, GeoVax will be able to enter into favorable manufacturing and distribution agreements, and other factors, over which GeoVax has no control.
Further information on our risk factors is contained in our periodic reports on Form 10-Q and Form 10-K that we have filed and will file with the SEC. Any forward-looking statement made by us herein speaks only as of the date on which it is made. Factors or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of them. We undertake no obligation to publicly update any forward-looking statement, whether as a result of new information, future developments or otherwise, except as may be required by law.
A Classic Since Always™ – From Revolutionary Pinsetters to Everyday Family Traditions
RICHMOND, Va.–(BUSINESS WIRE)– Lucky Strike Entertainment (NYSE: LUCK) today announced the launch of a refreshed identity for AMF (American Machine & Foundry), one of the most iconic names in American bowling. Rooted in AMF’s 126-year heritage, the rebrand positions the brand for the future and reflects its evolution into a modern bowling destination where families and communities come together for birthday parties, league nights, and social outings.
AMF’s refreshed shield logo marks a new chapter for an American classic.
Founded in 1900 as American Machine & Foundry, AMF helped shape modern bowling through innovations, including the automatic pinsetter and the world’s first automatic scoring system. Over the decades, the brand played a significant role in expanding bowling’s popularity across the United States and around the world, helping establish it as one of America’s most participated-in sports. Today, AMF remains a cornerstone of that legacy, welcoming approximately 10.4 million guests annually across more than 80 locations nationwide. With approximately 60 locations planned to transition to the AMF brand, Lucky Strike Entertainment is investing in the brand’s future while building on the foundation that made it an icon of the sport.
The AMF rebrand reflects Lucky Strike Entertainment’s vision to reintroduce AMF as a welcoming “house” with a refreshed identity that still honors the beloved sport. Rooted in AMF’s heritage, AMF destinations will begin to transform with bold new visuals led by Department of Branding and Design (DoBad) that lean into the brand’s ownable red, complemented by supporting tones of varsity blue, heritage white, and gold. The iconic shield logo will be updated with new lettering and bright red hues that will be reflected across digital and physical touchpoints. Consumers will be introduced to the new AMF brand platform, A Classic Since Always™, which celebrates modern bowling experiences while preserving the familiar, dependable atmosphere guests expect. Altogether, these elements position AMF as a timeless neighborhood destination designed to be the top bowling hub in neighborhoods across the country.
“This is more than a brand refresh. It’s an investment in the future of AMF,” said Thomas Shannon, Founder and CEO of Lucky Strike Entertainment. “We are building on the strength of an iconic brand and positioning it for long-term growth. As we expand the AMF brand across the country, our focus remains the same: delivering great bowling experiences, supporting league play, and providing an affordable destination for families and communities.”
Guests will begin to see the new AMF brand across digital platforms, social media, and select in-center materials, with additional updates rolling out over time. Future enhancements will include heritage walls showcasing memorabilia from AMF’s bowling legacy, along with nostalgic, varsity-inspired design elements such as swallowtail pennants and signage. While the look evolves, Lucky Strike Entertainment remains committed to AMF’s high-quality lanes, welcoming atmosphere, and experiences that bring people together, including leagues, open play, youth programs, family celebrations, and premier PBA Tour events such as the Tournament of Champions and AMF PBA World Championships. AMF also continues to offer affordable, value-driven birthday party options, with packages like Family Unlimited and the All Star Package designed to make celebrations easy, convenient, and accessible for families.
As AMF enters its next chapter, Lucky Strike Entertainment remains focused on elevating the traditional neighborhood bowling center, rooted in heritage, shaped by the local community, and designed to create moments of connection for generations to come.
For more information on AMF and AMF locations near you, please visit AMF.com and follow AMF on Instagram, and Facebook.
About Lucky Strike Entertainment
Lucky Strike Entertainment is one of the world’s premier location-based entertainment platforms. With over 360 locations across North America, Lucky Strike Entertainment provides experiential offerings in bowling, amusements, water parks, and family entertainment centers. The company also owns the Professional Bowlers Association, the major league of bowling and a growing media property that boasts millions of fans around the globe. For more information on Lucky Strike Entertainment, please visit LuckyStrikeEnt.com.
PDF VersionKratos to provide Mobile Counter-Unmanned Aircraft System (C-UAS) Platforms Designed to Support Critical National Security Mission
SAN DIEGO, July 21, 2026 (GLOBE NEWSWIRE) — Kratos Defense & Security Solutions, Inc. (Nasdaq: KTOS), a technology company in the defense, national security and global markets, today announced it has been awarded a sole-source, single award Indefinite Delivery/Indefinite Quantity (IDIQ) contract for approximately $156 million, by the U.S. Department of Energy’s National Nuclear Security Administration (NNSA) Office of Secure Transportation (OST), in support of Project Solar Shield.
Under this new contract award, Kratos will provide mobile Counter-Unmanned Aircraft System (C-UAS) platforms designed to support OST’s critical National Security mission. The Office of Secure Transportation is responsible for the safe and secure ground and air transportation of nuclear weapons, weapon components, and special nuclear materials, as well as other missions supporting U.S. national security.
To address emerging threats to these operations, OST requires a mission-ready mobile platform capable of detecting, tracking, identifying, and responding to potentially hostile unmanned aircraft systems in real time. Unlike traditional fixed-site defense infrastructure, Project Solar Shield is designed to provide a dynamic mobile C-UAS capability that can support mission requirements wherever OST operations occur. The program will leverage commercial and government best-in-class C-UAS technologies, to provide a layered defense posture supporting OST personnel and mission requirements.
Kratos was selected following a rigorous technical evaluation and was identified as the provider capable of meeting OST’s technical, cost, schedule, operational, safety, redundancy, integration, and long-term sustainment requirements. Kratos’ engineer-to-order approach combines C-UAS system, mobile platform design, command-and-control, and advanced power management technologies into a fully integrated solution, built for demanding threat and mission environments.
“Project Solar Shield represents a significant milestone for Kratos and the broader government C-UAS market,” said Dave Carter, President of Kratos’ Defense & Rocket Support Services Division. “As the first large scale government production contract of its kind built around this integrated approach, the program demonstrates the value of combining C-UAS, mobile platform design, command-and-control capabilities, and resilient power management into a single mission-ready solution. The resulting platform provides OST with a highly modular and scalable mobile C-UAS capability, designed to support evolving mission requirements across the continental United States.”
Eric DeMarco, President and CEO of Kratos, said, “We believe that Kratos’ technology, system and integration capabilities in the C-UAS mission area are industry leading, and also our ability to mass produce large quantities of relevant systems at an affordable cost. Our entire organization is proud to have received this program award to protect and secure critical United States assets and infrastructure.”
Work under this new program award will be performed at secure Kratos facilities. Due to security-related and other considerations, no additional information will be provided related to this contract award. Work under the contract is expected to begin immediately.
About Kratos Defense & Security Solutions Kratos Defense & Security Solutions, Inc. (NASDAQ: KTOS) is a technology, products, system and software company addressing the defense, national security, and commercial markets. Kratos makes true internally funded research, development, capital and other investments, to rapidly develop, produce and field solutions that address our customers’ mission critical needs and requirements. At Kratos, affordability is a technology, and we seek to utilize proven, leading-edge approaches and technology, not unproven bleeding edge approaches or technology, with Kratos’ approach designed to reduce cost, schedule and risk, enabling us to be first to market with cost effective solutions. We believe that Kratos is known as an innovative disruptive change agent in the industry, a company that is an expert in designing products and systems up front for successful rapid, large quantity, low-cost future manufacturing which is a value-add competitive differentiator for our large traditional prime system integrator partners and also to our government and commercial customers. Kratos intends to pursue program and contract opportunities as the prime or lead contractor when we believe that our probability of win (PWin) is high and any investment required by Kratos is within our capital resource comfort level. We intend to partner and team with a large, traditional system integrator when our assessment of PWin is greater or required investment is beyond Kratos’ comfort level. Kratos’ primary business areas include virtualized ground systems for satellites and space vehicles including software for command & control (C2) and telemetry, tracking and control (TT&C), jet powered unmanned aerial drone systems, hypersonic vehicles and rocket systems, propulsion systems for drones, missiles, loitering munitions, supersonic systems, space craft and launch systems, C5ISR and microwave electronic products for missile, radar, missile defense, space, satellite, counter UAS, directed energy, communication and other systems, and virtual & augmented reality training systems for the warfighter. For more information, visit www.KratosDefense.com and follow Kratos on LinkedIn and X.
Notice Regarding Forward-Looking Statements Certain statements in this press release may constitute “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements are made on the basis of the current beliefs, expectations and assumptions of the management of Kratos and are subject to significant risks and uncertainty. Investors are cautioned not to place undue reliance on any such forward-looking statements. All such forward-looking statements speak only as of the date they are made, and Kratos undertakes no obligation to update or revise these statements, whether as a result of new information, future events or otherwise. Although Kratos believes that the expectations reflected in these forward-looking statements are reasonable, these statements involve many risks and uncertainties that may cause actual results to differ materially from what may be expressed or implied in these forward-looking statements. For a further discussion of risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of Kratos in general, see the risk disclosures in the Annual Report on Form 10-K of Kratos for the year ended December 28, 2025, and in subsequent reports on Forms 10-Q and 8-K and other filings made with the SEC by Kratos.
Capital-efficient strategy focuses on differentiated therapies for cardiac pre- and post-operative critical care and orphan cardiovascular conditions
Company is exploring development, licensing, and commercialization partnerships for CAD-1005, frunexian, and tecarfarin
Recently presented Phase 2 CAD-1005 data demonstrated a >25% absolute reduction in thrombotic events in patients with heparin-induced thrombocytopenia
PONTE VEDRA, Fla., July 21, 2026 (GLOBE NEWSWIRE) — Cadrenal Therapeutics, Inc. (Nasdaq: CVKD), a late-stage biopharmaceutical company advancing specialized therapies for critical care cardiology and orphan cardiovascular conditions, today announced a comprehensive alignment of its clinical portfolio into a Cardiac Acute Critical Care Franchise. Concurrently, the Company has initiated a structured process to secure strategic out-licensing, portfolio monetization, or commercial co-development partnerships for its late-stage assets.
Cadrenal has assembled multiple late-stage critical care cardiovascular assets designed to address serious conditions for which existing treatment options remain inadequate. This capital-efficient operational model addresses a large, unserved therapeutic whitespace, unlocking substantial cumulative platform potential. The strategy positions Cadrenal’s assets as high-value additions which may be capable of delivering immediate value to the pipelines of global companies across rare disease therapies, critical care cardiology, and cardiac intensive care unit (CICU) medical products.
The strategic alignment of the Cardiac Acute Critical Care Franchise organizes Cadrenal’s portfolio into three high-value commercial pillars:
Pillar 1: Pre-Operative Safety (Frunexian IV): An IV Factor XIa inhibitor for heparin-induced thrombocytopenia (HIT)-susceptible patients undergoing coronary artery bypass graft (CABG) surgery, designed to replace unpredictable alternative protocols. Phase 2-ready.
Pillar 2: Orphan Regulatory Acceleration (including Tecarfarin for Kawasaki Disease): Leveraging substantial commercial tailwinds, fee waivers, and potential seven-year market exclusivity associated with the potential Orphan Drug Designation (ODD) pathways for cardiac surgery patients. Phase 3-ready.
Pillar 3: Post-Operative Shield (CAD-1005): A Phase 3-ready IV 12-LOX inhibitor designed to target platelet hyperactivation and thrombotic risk in patients with post-operative Heparin-Induced Thrombocytopenia (HIT). Secondary benefits may include blocking the inflammatory 12-HETE cascade to reduce cardiac surgery-associated acute kidney injury (CSA-AKI).
This franchise framework is strongly supported by the impressive clinical data presented earlier this month at the International Society on Thrombosis and Haemostasis (ISTH) 2026 Congress in Paris. The late-breaking Phase 2 data for CAD-1005 demonstrated a compelling medical profile, with a greater than 25% absolute reduction in thrombotic events and a favorable safety and renal-protective baseline.
“Our landmark clinical data presentation at ISTH 2026 has validated the mechanisms and the immense medical value of our Cardiac Acute Critical Care Franchise,” said Quang X. Pham, Chief Executive Officer of Cadrenal Therapeutics. “By organizing our pipeline into three distinct commercial pillars, we are positioning Cadrenal to maximize asset value. Transitioning to a partnership-driven business model enables us to map a direct, efficient path to commercialization alongside global industry leaders.”
In tandem with this strategic focus, the Company is advancing updates to its clinical pipeline for tecarfarin, highlighting its distinct carboxylesterase-1 (CES-1)-mediated metabolism, which helps avoid dangerous drug-drug interactions. This includes expanding clinical protocols to rare pediatric disease indications, such as giant coronary artery aneurysms (CAAs) caused by Kawasaki Disease-potentially qualifying the asset for a high-value, open-market Priority Review Voucher (PRV)-while continuing to support established indications in End-Stage Kidney Disease (ESKD) and Left Ventricular Assist Devices (LVADs).
Beyond the Cardiac Acute Critical Care Franchise, Cadrenal is advancing its preclinical platform asset, CAD-2000, a highly selective, orally bioavailable 12-lipoxygenase (12-LOX) inhibitor designed to treat chronic cardiorenal inflammatory and thrombotic indications. CAD-2000 serves as a potent oral follow-on companion to the Company’s IV acute care platform, potentially enabling prospective partners to capture extended market share across both acute inpatient settings and chronic outpatient follow-up care.
About CAD-1005
CAD-1005 is a novel investigational therapeutic in development for the treatment of heparin-induced thrombocytopenia (HIT). CAD-1005 is designed to selectively inhibit 12-lipoxygenase (12-LOX), an enzyme central to platelet immune activation and thrombo-inflammatory signaling in HIT. CAD-1005 is intended to be used alongside existing standards of care and is being developed to address the underlying biological mechanisms that drive disease progression. CAD-1005 has received Orphan Drug and Fast Track designations from the U.S. Food and Drug Administration and orphan drug status from the European Medicines Agency. Second-generation 12-LOX oral therapeutics are also in development for chronic indications. To view how CAD-1005 is intended to work in patients with HIT, click https://vimeo.com/1209382706/7dde06dc08?share=copy&fl=sv&fe=ci
About Cadrenal Therapeutics, Inc.
Cadrenal Therapeutics, Inc. is a late-stage biopharmaceutical company advancing specialized therapies for critical care cardiology and orphan cardiovascular conditions. Its lead program, CAD-1005, is being investigated as a first-in-class 12-LOX inhibitor for heparin-induced thrombocytopenia (HIT), a deadly immune-mediated thrombotic disorder. The Company’s Cardiac Acute Critical Care Franchise also includes frunexian, an investigational intravenous Factor XIa inhibitor designed to provide anticoagulation for patients undergoing major cardiac surgery.
The Company’s broader pipeline includes tecarfarin, a late-stage oral vitamin K antagonist designed to prevent heart attacks, strokes, and deaths from blood clots in patients requiring chronic anticoagulation, including those with end-stage kidney disease, those with left ventricular assist devices, and potentially those with Kawasaki disease (KD), an acute, self-limited, febrile illness that primarily affects children under 5 years old and is the leading cause of acquired heart disease in developed countries. Tecarfarin has also received Orphan Drug and Fast Track designations from the U.S. Food and Drug Administration (FDA).
Safe Harbor
Any statements in this press release about future expectations, plans, and prospects, as well as any other statements regarding matters that are not historical facts, may constitute “forward-looking statements.” The words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potentially,” “predict,” “project,” “should,” “target,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. These statements include, without limitation, statements such as Cadrenal launching a partnering process; advancing its Cardiac Acute Critical Care Assets to transaction readiness following landmark ISTH data presentation; the Company’s capital-efficient strategy focusing on differentiated therapies for cardiac pre- and post-operative critical care and orphan cardiovascular conditions; the Company exploring development, licensing, and commercialization partnerships for CAD-1005, frunexian, and tecarfarin; the Company advancing specialized therapies for critical care cardiology and orphan cardiovascular conditions; the Company securing strategic out-licensing, portfolio monetization, or commercial co-development partnerships for its late-stage assets; Cadrenal’s multiple late-stage critical care cardiovascular assets addressing serious conditions for which existing treatment options remain inadequate; the capital-efficient operational model addressing a large, unserved therapeutic whitespace, unlocking substantial cumulative platform potential; Cadrenal’s assets delivering immediate value to the pipelines of global companies across rare disease therapies, critical care cardiology, and cardiac intensive care unit (CICU) medical products; frunexian IV successfully replacing unpredictable alternative protocols; the potential of CAD-1005 to block the inflammatory 12-HETE cascade to reduce cardiac surgery-associated acute kidney injury; positioning Cadrenal to maximize asset value; transitioning to a partnership-driven business model enabling the Company to map a direct, efficient path to commercialization alongside global industry leaders; the Company advancing updates to its clinical pipeline for tecarfarin, highlighting its distinct carboxylesterase-1 (CES-1)-mediated metabolism, which helps avoid dangerous drug-drug interactions; expanding clinical protocols to rare pediatric disease indications, such as giant coronary artery aneurysms (CAAs) caused by Kawasaki Disease-potentially qualifying the asset for a high-value, open-market Priority Review Voucher (PRV)-while continuing to support established indications in End-Stage Kidney Disease (ESKD) and Left Ventricular Assist Devices (LVADs); Cadrenal advancing its preclinical platform asset, CAD-2000, a highly selective, orally bioavailable 12-lipoxygenase (12-LOX) inhibitor designed to treat chronic cardiorenal inflammatory and thrombotic indications. CAD-2000 serves as a potent oral follow-on companion to the Company’s IV acute care platform, potentially enabling prospective partners to capture extended market share across both acute inpatient settings and chronic outpatient follow-up care; tecarfarin potentially treating patients with Kawasaki disease (KD). Actual results may differ materially from those indicated by such forward-looking statements as a result of various important factors, including the ability to enter into a partnership opportunity, development, licensing and commercialization for CAD-1005 frunexian, and tecarfarin; the ability of Cadrenal’s assets to be additions to the pipelines of global companies across rare disease therapies, critical care cardiology, and cardiac intensive care unit medical products; the ability to benefit from the commercial potential of the Cadrenal’s product candidates; the ability to raise sufficient capital to continue the clinical development of its product candidates; and the other risk factors described in the Company’s Annual Report on Form 10-K for the year ended December 31, 2025, and the Company’s subsequent filings with the Securities and Exchange Commission, including subsequent periodic reports on Quarterly Reports on Form 10-Q and Current Reports on Form 8-K. Any forward-looking statements contained in this press release speak only as of the date hereof and, except as required by federal securities laws, the Company specifically disclaims any obligation to update any forward-looking statement, whether as a result of new information, future events, or otherwise.
NEW ALBANY, Ohio, July 21, 2026 (GLOBE NEWSWIRE) — Commercial Vehicle Group (the “Company” or “CVG”) (NASDAQ: CVGI) will hold its quarterly conference call on Tuesday, August 4, 2026, at 8:30 a.m. ET, to discuss second quarter 2026 financial results. CVG will issue a press release and presentation prior to the conference call.
Toll-free participants dial (833) 461-5787 using conference code 592968497. International participants dial (585) 542-9983 using conference code 592968497. This call is being webcast and can be accessed through the “Investors” section of CVG’s website at ir.cvgrp.com where it will be archived for one year.
About CVG
Commercial Vehicle Group, Inc. and its subsidiaries, is a global provider of systems, assemblies and components to global commercial vehicle markets and electric vehicle markets. We deliver real solutions to complex design, engineering and manufacturing problems while creating positive change for our customers, industries, and communities we serve. Information about the Company and its products is available on the internet at www.cvgrp.com.
Investor Relations Contact: Ross Collins or Nathan Skown Alpha IR Group [email protected]
CHICAGO, July 21, 2026 (GLOBE NEWSWIRE) — FreightCar America, Inc. (NASDAQ: RAIL) (“FreightCar America” or the “Company”), a diversified manufacturer and supplier of railroad freight cars, railcar parts and components, today announced that it has completed the acquisition of Southern Parts & Equipment, Inc. (“SP&E”), a longstanding distributor of new, used and reconditioned railcar parts and equipment. The transaction marks the Company’s second acquisition in the railcar aftermarket space within the past year.
The acquisition further expands FreightCar America’s aftermarket distribution capabilities and strengthens the Company’s ability to serve customers with a broader offering of railcar components and related services. Southern Parts & Equipment has built a long-standing presence in the rail industry, supplying railcar parts and equipment and supporting customers with rail industry consulting, inspections and project-related services.
“Southern Parts & Equipment is a highly complementary addition to our growing aftermarket platform,” said Nicholas Randall, President and Chief Executive Officer of FreightCar America. “This acquisition further strengthens our ability to support customers with improved access to critical railcar components, while advancing our strategy to build complementary aftermarket capabilities that increase the value we deliver across the railcar lifecycle.”
“We are excited to welcome Southern Parts & Equipment to FreightCar America,” said Michael Riordan, Vice President, Chief Financial Officer & Treasurer of FreightCar America. “Southern Parts & Equipment brings valuable customer relationships, product knowledge and sourcing capabilities that align well with our aftermarket growth strategy. By combining its capabilities with FreightCar America’s commercial reach, supply chain discipline and growing aftermarket platform, we believe we can deliver greater value to customers while continuing to scale a more diversified and recurring revenue stream. This transaction is consistent with our disciplined capital allocation framework and is expected to be immediately accretive to FreightCar America.”
About Southern Parts & Equipment
Founded in Monroe, Georgia, Southern Parts & Equipment supplies new, used and reconditioned railcar parts and equipment to the railroad industry. The company serves repair shops, railroads, private car owners and other industrial customers, and has earned a decades-long reputation as a trusted source for railcar components. To learn more about Southern Parts & Equipment, visit www.southernrailparts.com.
AboutFreightCarAmerica
FreightCar America, headquartered in Chicago, Illinois, is a leading designer, producer and supplier of railroad freight cars, railcar parts and components. We also specialize in railcar repairs, complete railcar rebody services and railcar conversions that repurpose idled rail assets back into revenue service. Since 1901, our customers have trusted us to build quality railcars that are critical to economic growth and instrumental to the North American supply chain. To learn more about FreightCar America, visit www.freightcaramerica.com.
Forward-Looking Statements
This press release may contain statements relating to our expected financial performance, financial condition, and/or future business prospects, events and/or plans that are “forward-looking statements” as defined under the Private Securities Litigation Reform Act of 1995. Forward-looking statements represent our estimates and assumptions only as of the date of this press release. Our actual results may differ materially from the results described in or anticipated by our forward-looking statements due to certain risks and uncertainties. These risks and uncertainties relate to, among other things, the cyclical nature of our business; adverse geopolitical, economic and market conditions, including inflation; material disruption in the movement of rail traffic for deliveries; fluctuating costs of raw materials, including steel and aluminum; delays in the delivery of raw materials; our ability to maintain relationships with our suppliers of railcar components; our reliance upon a small number of customers that represent a large percentage of our sales; the variable purchase patterns of our customers and the timing of completion; delivery and customer acceptance of orders; the highly competitive nature of our industry; the risk of lack of acceptance of our new railcar offerings; potential unexpected changes in laws, rules, and regulatory requirements, including tariffs and trade barriers (including recent United States tariffs imposed or threatened to be imposed on China, Canada, Mexico and other countries and any retaliatory actions taken by such countries); and other competitive factors. The factors listed above are not exhaustive. New factors emerge from time to time that may cause our business not to develop as we expect, and it is not possible for us to predict all of them. We expressly disclaim any duty to provide updates to any forward-looking statements made in this press release, whether as a result of new information, future events or otherwise.
ITS Designs and Delivers a Custom Composite Materials Production Line, Extending Defense-Grade Fiber-Reinforced Composite Manufacturing to Industrial Scale
NEW YORK and NETANYA, Israel, July 20, 2026 (GLOBE NEWSWIRE) — T3 Defense Inc. (Nasdaq: DFNS) (“T3 Defense” or the “Company”), a defense company that acquires and operates mission-critical defense and industrial businesses, today announced that its subsidiary, Industrial Techno-Logic Solutions (“ITS”), has designed and delivered a custom composite materials production line to a leading Israeli building materials manufacturer.
Bringing its defense-proven fiberglass production expertise to a line for a customer in building materials, ITS was able to fully integrate automated materials handling, precision application, and fiberglass-based reinforcement to manufacture advanced composite products at industrial scale. Applying its Design for Manufacturing (DFM) methodology, ITS assumed end-to-end responsibility for mechanical engineering design, machining, supply chain management, integration, and factory deployment, enabling the manufacturer to achieve production-ready output with the precision and delivery reliability that characterizes ITS’s work across defense and industrial programs.
“Defense capability increasingly depends on sovereign manufacturing supply chains. Nations that cannot produce the components their defense programs require at home face critical vulnerabilities at exactly the wrong moment. ITS is closing that gap,” said Menny Shalom, Chairman and CEO of T3 Defense. “Composite materials are becoming foundational across defense and industrial manufacturing, and the ability to produce them reliably at scale is a strategic capability in its own right. This production line gives the customer exactly that foundation, at the precision that defense and industrial programs demand. As governments and prime contractors continue to prioritize localized, qualified manufacturing, we expect ITS to be an increasingly sought-after partner for exactly this type of capability-building engagement.”
The delivery marks the latest milestone in an ongoing relationship between ITS and the undisclosed customer, reflecting confidence the customer has placed in ITS as a long-term manufacturing partner. The expansion of the collaboration to include a dedicated continuous production line underscores ITS’s ability to support customers across successive programs and scale, translating early-stage engineering partnerships into full production-ready industrial infrastructure.
About ITS
Industrial Techno-Logic Solutions (ITS) is an engineering and manufacturing systems company that develops specialized production lines for complex defense and industrial technologies. Using a Design for Manufacturing (DFM) methodology, ITS designs production systems that enable advanced products to be manufactured reliably and at scale. By combining engineering design, machining, supply chain management, and factory deployment, ITS helps customers move technologies from concept to production-ready manufacturing environments. ITS is a majority-owned subsidiary of T3 Defense Inc. For more information, visit www.its-eng.com.
About T3 Defense
T3 Defense Inc. (Nasdaq: DFNS) is a defense company that acquires and operates mission-critical defense businesses embedded in long-cycle national security programs. The company targets businesses operating at constrained, qualification-driven, or execution-critical points across the industrial base where strategic value exists and where qualification, capacity, and execution are decisive. Through disciplined M&A, centralized capital and strategy, and decentralized operating autonomy, T3 Defense seeks to strengthen critical defense capabilities and compound long-term value. For more information, visit www.t3dfns.com.
Forward Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements include statements regarding ITS’s engineering and manufacturing capabilities, the expected performance of the production line delivered to the client, and the Company’s growth strategy. These statements involve known and unknown risks and uncertainties that could cause actual results to differ materially from those expressed or implied. T3 Defense Inc. undertakes no obligation to update or revise any forward-looking statements to reflect events or circumstances after the date of this press release, except as required by applicable law.
ATLANTA, July 20, 2026 (GLOBE NEWSWIRE) — DLH Holdings Corp. (NASDAQ: DLHC) (“DLH” or the “Company”), a leading provider of science research and development, systems engineering and integration, and digital transformation and cyber security solutions to federal agencies, will release financial results for the fiscal third quarter ended June 30, 2026 on July 29, 2026 after the market closes. DLH will then host a conference call for the investment community at 10:00 a.m. Eastern Time the following day, July 30, 2026, during which members of senior management will make a brief presentation focused on the financial results and operating trends. A question-and-answer session will follow.
Interested parties may listen to the conference call by dialing 888-347-5290 or 412-317-5256. Presentation materials will also be posted on the Investor Relations section of the DLH website prior to the commencement of the conference call. A digital recording of the conference call will be available for replay two hours after the completion of the call and can be accessed on the DLH Investor Relations website or by dialing 1-855-669-9658 and entering the conference ID 1652291.
About DLH DLH (NASDAQ: DLHC) enhances technology, public health, and cyber security readiness missions through science, technology, cyber, and engineering solutions and services. Our experts solve some of the most complex and critical missions faced by federal customers, leveraging digital transformation, artificial intelligence, advanced analytics, cloud-based applications, telehealth systems, and more. With a world-class workforce dedicated to the idea that “Your Mission is Our Passion,” DLH brings a unique combination of government sector experience, proven methodology, and unwavering commitment to innovative solutions to improve the lives of millions. For more information, visit www.DLHcorp.com.
INVESTOR RELATIONS Contact: Chris Witty Phone: 646-438-9385 Email: [email protected]
Elroy Air Recently Announced a Demand Pipeline Exceeding 1,400 Aircraft
Kratos to Increase Current Sacramento Workforce of 450+ High-Tech Employees as Production of Elroy Air’s Autonomous Cargo Aircraft Accelerates
SAN DIEGO, July 20, 2026 (GLOBE NEWSWIRE) — Kratos Defense & Security Solutions, Inc. (NASDAQ: KTOS), a Technology Company in the Defense, National Security and Global Markets, today announced that it will manufacture Elroy Air’s Chaparral autonomous cargo aircraft in its expanding Sacramento, California production facility, supporting increasing demand across commercial logistics and defense markets while expecting to further grow its regional workforce of 450 high-tech employees by more than 50 as Chaparral production ramps.
The Chaparral is a hybrid-electric, vertical takeoff and landing (VTOL) autonomous cargo aircraft designed to transport more than 500 pounds of payload with a maximum range of up to 450 miles without requiring traditional airport infrastructure. The system is designed to support commercial middle-mile logistics while also providing a flexible, autonomous resupply capability for military operations.
The announcement marks the transition from strategic manufacturing partner to production execution following Elroy Air’s recent announcement of its planned public listing and continued commercial momentum. Kratos is the exclusive U.S. manufacturer of the Chaparral aircraft and will fulfill all U.S. customer orders, with the first production aircraft planned for late 2026. Recent expansion of Kratos’ Sacramento manufacturing operations provides the production capacity necessary to support anticipated increases in aircraft deliveries.
Located within driving distance of Elroy Air’s headquarters, the expanded Sacramento facility strengthens collaboration between the two companies while increasing manufacturing capacity for one of the industry’s most advanced autonomous cargo aircraft. The expansion will drive additional hiring across aircraft technicians, composite manufacturing specialists, assemblers, engineers, production operations, quality assurance, and program management positions, bringing Kratos’ Sacramento-area workforce to more than 500 employees.
Steve Fendley, President of Kratos’ Unmanned Systems Division, said, “At Kratos, we have built our business around rapidly transitioning advanced unmanned aircraft from development into affordable, scalable production. Chaparral represents another example of Kratos leveraging its proven manufacturing capability, established supply chain, and experienced workforce to help bring an innovative aircraft into production at scale. As demand continues to build, our expanding Sacramento facility is well positioned to support both commercial and defense customers while creating additional high-value aerospace jobs in California.”
Dr. Andrew Clare, CEO of Elroy Air, said, “Demand for Chaparral is accelerating across defense, rapid response and commercial logistics and meeting it requires manufacturing at scale. Partnering with Kratos lets us build American-made autonomous cargo drones right here in California, at the pace our customers need.”
Elroy Air recently announced a demand pipeline exceeding 1,400 aircraft representing more than $5 billion in potential revenue opportunities from leading logistics and aviation companies, including Bristow Group, Barq Group, SLI, and FedEx. The company has also supported defense programs with the U.S. Army, U.S. Marine Corps, and U.S. Air Force for more than six years, demonstrating the growing dual-use market opportunity for the Chaparral platform. The company also recently announced plans to become a publicly traded company, positioning it to accelerate commercial-scale production.
Kratos continues to expand its national manufacturing footprint to meet increasing demand for affordable, mission-ready unmanned systems supporting U.S. and allied defense priorities, while enabling the production of innovative dual-use technologies serving both commercial and government customers.
About Kratos Defense & Security Solutions Kratos Defense & Security Solutions, Inc. (NASDAQ: KTOS) is a technology, products, system and software company addressing the defense, national security, and commercial markets. Kratos makes true internally funded research, development, capital and other investments, to rapidly develop, produce and field solutions that address our customers’ mission critical needs and requirements. At Kratos, affordability is a technology, and we seek to utilize proven, leading-edge approaches and technology, not unproven bleeding edge approaches or technology, with Kratos’ approach designed to reduce cost, schedule and risk, enabling us to be first to market with cost effective solutions. We believe that Kratos is known as an innovative disruptive change agent in the industry, a company that is an expert in designing products and systems up front for successful rapid, large quantity, low-cost future manufacturing which is a value-add competitive differentiator for our large traditional prime system integrator partners and also to our government and commercial customers. Kratos intends to pursue program and contract opportunities as the prime or lead contractor when we believe that our probability of win (PWin) is high and any investment required by Kratos is within our capital resource comfort level. We intend to partner and team with a large, traditional system integrator when our assessment of PWin is greater or required investment is beyond Kratos’ comfort level. Kratos’ primary business areas include virtualized ground systems for satellites and space vehicles including software for command & control (C2) and telemetry, tracking and control (TT&C), jet powered unmanned aerial drone systems, hypersonic vehicles and rocket systems, propulsion systems for drones, missiles, loitering munitions, supersonic systems, space craft and launch systems, C5ISR and microwave electronic products for missile, radar, missile defense, space, satellite, counter UAS, directed energy, communication and other systems, and virtual & augmented reality training systems for the warfighter. For more information, visit www.KratosDefense.com and follow Kratos on LinkedIn and X.
Notice Regarding Forward-Looking Statements Certain statements in this press release may constitute “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements are made on the basis of the current beliefs, expectations and assumptions of the management of Kratos and are subject to significant risks and uncertainty. Investors are cautioned not to place undue reliance on any such forward-looking statements. All such forward-looking statements speak only as of the date they are made, and Kratos undertakes no obligation to update or revise these statements, whether as a result of new information, future events or otherwise. Although Kratos believes that the expectations reflected in these forward-looking statements are reasonable, these statements involve many risks and uncertainties that may cause actual results to differ materially from what may be expressed or implied in these forward-looking statements. For a further discussion of risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of Kratos in general, see the risk disclosures in the Annual Report on Form 10-K of Kratos for the year ended December 28, 2025, and in subsequent reports on Forms 10-Q and 8-K and other filings made with the SEC by Kratos.
PDF Version New capabilities position Company to pursue additional global defense opportunities
CHARLOTTE, N.C., July 20, 2026 (GLOBE NEWSWIRE) — NN, Inc. (NASDAQ: NNBR) (“NN” or the “Company”), a global diversified industrial company that engineers, co-develops and manufactures precision components and assemblies with six sigma quality, today announced entry into a brand-new market segment for the company. The company has successfully entered the Tier 1 contract manufacturing industry for firearm components in the United States market. The key components of this successful market entry are:
Turnkey Tier 1 contract manufacturing
Collaborative product development
Additive manufacturing during prototyping
High volume titanium machining
High volume laser welding and assembly
Multiple surface treatment advancements including ceramic surface coating, physical vapor deposition, and nanocomposite diamond-like carbon coating
ATF and FFL compliance program
CMMC Tier 2 certification program
TISAX Tier 2 certification program
ITAR compliance program
Encrypted communications portal that is CMMC, NIST, ITAR and HIPAA compliant
Attendance at weapons shows in the United States
NN has entered into a contract manufacturing agreement to mass produce completed firearms products for a leading provider of firearms products in the United States. This new business for NN was achieved after a multi-year effort that required:
New products
Titanium machining breakthroughs
Surface treatment breakthroughs
Specialized equipment investments
Multi-year collaborative innovation program with a leading brand owner
This new product category is included within NN’s business growth program in Defense & Electronics. This new business begins in Q3 and will continue ramping up through 2028. This new business is expected to add between $12 million to $15 million in sales.
Growth in Defense & Electronics is a key component of the company’s 5-point growth plan. It is a direct application and natural extension of its in-house capabilities into a new market. Including these new wins, NN has won 20+ new programs worth >$30 to $35 million per year over the last 3 years. The company also has an additional pipeline in Defense & Electronics of $75 million of opportunities and has recently hired a Defense industry specialist. Of note, the company is evaluating the manufacturing of munitions for attack drones.
NN has a multi-product game plan within its Defense & Electronics growth program.
Gold and silver plating of critical electronics modules that are contained in advanced weapon systems
Complicated metal fabrications that are key components within guidance and weapon systems
High-end machined parts that are components in firearms (the focus on this article)
The US defense market is at record spending levels with a 5-year outlook to keep growing. Specific to this new set of awards, the US firearms market is growing due to increased emphasis on baseline safety.
The company is employing a large portion of its US footprint to make products for the defense, electronics, and weapons markets. NN now has 8 plants that are ITAR compliant. This enables the company to have a wide aperture onto these markets with a broad product offering of machined parts, stamped parts, plated parts, and assemblies.
Harold Bevis, President and Chief Executive Officer of NN, Inc., commented, “This is a nice advancement for NN’s sales growth program into new markets. It is another direct payoff on the investments we are making to establish premier positions in high-value markets. A tremendous amount of collaborative innovation occurred between the brand owner and NN over the last couple of years on this program. Many prototypes and many samples were iterated in order to arrive at the perfect next-generation product performance.
“We are creating additional competitive barriers by adding distinguishing factory credentials for this new market – ATF and FFL compliance, ITAR compliance, CMMC Tier 2 certification, and TISAX Tier 2 certification. These credentials enhance participation in these markets.
“A fun fact that is due to the multipart complexity of this new product line is that these products are now the highest-priced products in the company’s portfolio of new products. Prices will range from $200 to $500 for each product.”
Bevis concluded, “NN is underway with a multi-year program of sales-driven earnings improvements and this is another building block. Our Defense products growth program is achieving victories. The Defense & Electronics business is already nearly $60 million in sales, and we have a 5-year goal for it to grow to $100 million. It is one of our most profitable segments also due to higher value-add. Our 5 pillars of entrepreneurial break-out growth continue to be focused upon: data center and electric grid, defense and electronics, medical products, high-value vehicle products, and high-value stamped products.
“We will combine this new information along with recent new wins in Data Center and Medical products and adjust 2026 guidance, if needed, for sales, adjusted EBITDA, and New Wins when we release Q2 2026 earnings on August 6. We look forward to discussing this new advancement further at that time.”
Suppressor Pieces and Assembled Product
About NN, Inc. NN, Inc., a global diversified industrial company, combines advanced engineering and production capabilities with in-depth materials science expertise to design and manufacture high-precision components and assemblies for a variety of markets on a global basis. Headquartered in Charlotte, North Carolina, NN has facilities in North America, South America, Europe, and China. For more information about the company and its products, please visit www.nninc.com.
Forward-Looking Statements This press release contains express and implied forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995, including, but not limited to, statements regarding the future growth of NN’s medical business, including NN’s expectations regarding current and future customers and programs, the size and future outlook of the medical market, including robotics-assisted surgery, NN’s competitive position in the medical market, expected new business wins for 2026, and NN’s 2026 performance and other statements that are not historical facts.
Forward-looking statements generally will be accompanied by words such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “forecast,” “guidance,” “intend,” “may,” “will,” “possible,” “potential,” “predict,” “project”, “achieve,” “growth,” “enable,” “improve,” or the negative of these terms, and similar words, phrases or expressions that convey uncertainty of future events or outcomes. Forward-looking statements involve a number of risks and uncertainties that are outside of management’s control and that may cause actual results to be materially different from such statements. Such factors include, among others, general economic conditions and economic conditions in the industrial sector; competitive influences; risks that current customers will commence or increase captive production; risks of capacity underutilization; quality issues; inflationary pressures and material changes in the costs and availability of raw materials, supply chain shortages and disruptions, the availability of labor and labor distributions along the supply chain; our dependence on certain major customers, some of whom are not parties to long-term agreements (and/or are terminable on short notice); the impact of acquisitions and divestitures, as well as expansion of end markets and product offerings; our ability to hire or retain key personnel; the restrictions contained in our debt agreements; the level of our indebtedness and our ability to financing at favorable rates, if at all, or to refinance existing debt as it matures; our ability to secure, maintain or enforce patents or other appropriate protections for our intellectual property; the impact on climate change on our operations; economic, social and geopolitical instability, military conflict, currency fluctuations, and other risks of doing business outside of the United States; and uncertainty of government policies and actions in respect to global trade and tariffs, including the potential impacts of tariffs on the United States economy, the economy of other countries in which we conduct operations and our industry, cyber liability or potential liability for breaches of our or our service providers’ information technology systems or business operations disruptions. The foregoing factors should not be construed as exhaustive and should be read in conjunction with the sections entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in the Company’s filings made with the U.S. Securities and Exchange Commission. Any forward-looking statement speaks only as of the date of this press release, and the Company undertakes no obligation to publicly update or review any forward-looking statement, whether as a result of new information, future developments or otherwise, except as required by law. New risks and uncertainties may emerge from time to time, and it is not possible for the Company to predict their occurrence or how they will affect the Company. The Company qualifies all forward-looking statements by these cautionary statements.
Investor & Media Contact: Joe Caminiti [email protected] 312-445-2870
STAFFORD, Texas, July 20, 2026 (GLOBE NEWSWIRE) — Greenwich LifeSciences, Inc. (Nasdaq: GLSI) (the “Company”), a clinical-stage biopharmaceutical company focused on its Phase III clinical trial, FLAMINGO-01, which is evaluating GLSI-100, an immunotherapy to prevent breast cancer recurrences, today provided the following clinical updates on FLAMINGO-01.
FLAMINGO-01 Data Safety Monitoring Board (DSMB)
The FLAMINGO-01 DSMB met in May 2026 and recommended the study continue as is without modification.
FLAMINGO-01 Steering Committee Clinical Strategy
On December 22, 2025, the company announced the following objectives:
“The Steering Committee also met at SABCS 2025 and discussed the clinical strategy, endorsing the planned modifications to FLAMINGO-01. The planned modifications subject to regulatory approval include:
increasing the size of the study, which would increase the power of the study thus decreasing the risk by designing the study to assume more recurrences even though fewer recurrences may be anticipated and observed,
doubling or quadrupling the enrollment rate, which will increase the patient years in the study more rapidly thus proportionately increase the event rate, which may shorten the time to reach an interim analysis or milestone,
continuing to enroll past the interim analyses so that the current momentum at the clinical sites continues,
using the interim analysis to potentially resize the study or to change the subsequent interim analysis, to change the number of events triggering an analysis, or to change the timing of the study based on recommendations by an independent committee, and
using a recently manufactured GP2 commercial drug product lot in FLAMINGO-01″
CEO Snehal Patel commented, “We are pleased to announce that following review by FDA and EMA regulatory authorities that these objectives have been met and that FLAMINGO-01 now has the hallmarks of a large pharma Phase III clinical trial. The study is now designed and sized to improve the probability of success, to attract the interest of sophisticated investors and pharma companies, and to maximize the chances that the Company could file a BLA after interim analysis 1, after interim analysis 2, or after the end of the study.”
Mr. Patel further added, “The transition is now underway globally at all sites. We have provided below in this press release the details of the study design reviewed by both the US and EU agencies, and currently subject to review by the UK and Canada, and will be updating the Company website and presentation, www.ClinicalTrials.gov, and videos accordingly. These agencies may provide additional recommendations or requirements at any time and we remain flexible to accommodate their advice as needed.”
The Company plans to now leverage the increased enrollment rate resulting from the combination of all HLA types together with the following: 1) the very high interest from patients and clinicians which has led to almost 200 clinical trial sites in the US and Europe, 2) the clinical operational capability in place 3) the currently trending low event rate, and 4) the efficient cost structure and burn rate that the Company has successfully funded through small capital raises.
Enrollment Rate into the Blinded Arm
As previously disclosed, all European and US Sites will combine all new patients independent of HLA type in the randomized arms of FLAMINGO-01. Non-HLA-A*02 patients who represent about 55% of the population and were on waiting lists for up to a year are now eligible for enrollment, which could provide for the rapid enrollment of up to 300 patients. This protocol amendment will more than double the enrollment rate increasing it by 122% or resulting in a 2.22x faster enrollment rate (55%/45% = 122%), which proportionately increases the event rate. This more than doubling of the event rate, provides an opportunity to derisk the study and provide multiple pathways to filing a BLA in the US with much higher probabilities of success at each opportunity for analysis.
Leveraging the High Interest from Patients and Clinicians
The Company has achieved a major milestone by screening over 1,500 patients in Flamingo-01, continuing its screening rate of approximately 150-200 patients per quarter or the equivalent of 600-800 patients per year in approximately 170-180 sites.
The 11 participating countries include: US, Spain, France, Germany, Italy, Poland, Romania, Ireland, Portugal, Belgium, and Austria. The Company is planning to add the following additional European countries due to interest from principal investigators and patients: Norway, Denmark, and Sweden in addition to the UK and Canada.
Rationale to Keep Enrollment Open Until Interim Analyses
In the double-blinded arms of the Phase III trial, the originally designed 500 HLA-A*02 patients were likely to be filled before the interim analysis and thus the clinical sites would have had to stop enrolling. If the interim analysis suggested that more patients should be enrolled, restarting enrollment would have been very difficult. A 2.22x increase in the enrollment rate without any other modifications would have accelerated the stopping of enrollment, but the probability of a successful data analysis at the interim analysis and the filing of BLA would not be increased. It isn’t in the study’s best interest to wait for more events without the option to continue enrolling to increase the event rate. It is optimal to keep enrolling while collecting events because even patients in the study for only a short time are at risk of recurrence and can add information to analyses.
Capital Raising Strategy Has Kept up with Modestly Increasing Burn Rate
The cash burn will be manageable as in the past due to the efficiently run and internalized clinical operations. The manufacturing of GP2 vials for the Phase III clinical trial has been completed with sufficient vials to treat all patients. Most of the start-up costs for the clinical sites have been paid. Many patients have entered the booster phase with 2 vaccinations per year with lower costs thus offsetting the higher costs for patients entering the study, when 6 vaccinations in the first 6 months during the primary immunization series are required.
The Company’s annual burn rate was approximately $7 million in 2024 and 2023 and $10 million in 2025. The income statements for these periods have been reported as much higher losses, but the cash flow used for operations is much lower due to the non-cash stock and options expenses added to the income statements.
For the second quarter of 2026, the burn rate is expected to be approximately $2 million versus a $4.7 million burn rate in the first quarter of 2026, leading to a Q2 2026 cash balance of approximately $8.9 million as of June 30, 2026 and an expected burn rate of $2-4 million per quarter going forward. The above preliminary financial figures are unaudited and are subject to change following completion of the Company’s financial review for Q2 2026. This capital raising strategy may provide a bridge to non-dilutive funding, such as strategic/licensing partnerships or debt/royalty financing vehicles, that would further fund FLAMINGO-01 and potential commercial launch activities.
Improved Trial Design Allows for Substantial Reduction in Risk
In the double-blinded arms of the Phase III trial, the trial has been designed to detect a hazard ratio (HR) of 0.55 in invasive breast cancer-free survival, where 28 events will be required for the 1st interim analysis, 56 events will be required for the 2nd interim analysis, and 133 events will be required to end the study. Interim analyses for superiority and futility will be conducted and, if successful, could lead to the filing of a BLA in the US at those times. The number of patients enrolled in the study will depend on the event rate and thus enrollment may continue for as long as necessary up to a maximum of 2,000 patients. This sample size provides 80% power if the annual rate of events in placebo-treated subjects is 2.4% or greater and the HR is 0.55. In addition, the number of events may be adapted by the DSMB based on interim analyses.
By doubling the number of events to trigger an interim and increasing the HR, which is offset by the more than doubling event rate due to the higher enrollment rate, and may or may not alter time lines, the probability of a positive study outcome can be increased substantially. An increase of the HR puts FLAMINGO-01 more in line with other prominent large pharma breast cancer Phase III clinical trials such as Katherine for Kadcyla and Destiny Breast-05 for Enhertu. The HR is equal to one minus the percent reduction in events caused by the treatment arm. For example, an HR = 0.3 would suggest a 70% reduction in events and an HR = 0.75 would suggest a 25% reduction in events by the treatment arm.
The Katherine study which compared Kadcyla to Herceptin breast cancer treatment in the adjuvant setting after surgery in the residual disease population assumed a design HR = 0.75 but realized a lower study result HR at the first interim of 0.5, which led to a sufficiently low p value and strong enough statistical significance to warrant approval for Kadcyla in the adjuvant setting following submission of interim data to the FDA. Approximately 1,486 patients were enrolled, 256 events were observed at the interim analysis, and 385 events were observed at the final analysis.
The Destiny Breast-05 study which compared Enhertu to Kadcyla breast cancer treatment in the adjuvant setting after surgery, in a higher risk residual disease population than Katherine, assumed a design HR = 0.675 but realized a lower study result HR at the first interim of 0.5, which led to a sufficiently low p value and strong enough statistical significance to warrant approval for Enhertu in the adjuvant setting following submission of interim data to the FDA. Approximately 1,635 patients were enrolled and 153 events were observed at the interim analysis.
GLSI-100 by contrast has shown a study result HR = 0.2 in the Phase IIb clinical trial for HLA-A*02 patients. In the 250 patient non-HLA-A*02 open label arm of FLAMINGO-01, which is now fully enrolled and where all patients received GLSI-100, a preliminary analysis of recurrence rates after the PIS is completed shows an approximately 70-80% reduction in recurrence rate or a HR = 0.2-0.3 when calculated by various methods. This data is early and will continue to mature over time. This low event rate is supported by immune response data that was recently published at AACR and ASCO conferences in 2026. The section below, “About FLAMINGO-01 Open Label Phase III Data”, summarizes these results and provides links to the posters at the conferences.
By increasing the original FLAMINGO-01 trial design HR = 0.3 to a design HR = 0.55 and by doubling the events required to trigger the first interim analysis from 14 to 28 events, the probability of success or power at the first interim analysis, if a lower study result HR = 0.3 is realized, increases from less than 20% to more than 85%. This increase in power at the first interim, when the study result HR is lower than the design HR, is possible due to the combination of both HLA types, while the more than double event rate at 2.22x offsets the doubling of the events required to trigger this first interim analysis. Effectively increasing the probability of filing a BLA at the first interim analysis from 20% to 85% justifies the study design changes.
Additional benefits of the new design include:
The first interim results may affect or alter the design of the second interim.
Endpoints can be analyzed by individual HLA types as well as one combined group of all HLA types and given that each patient has 2 HLA-A alleles, one from each parent, there are multiple analyses that can be conducted.
Doubling the market for GLSI-100 to potentially $10 billion in revenue per year by accelerating the clinical development of the non-HLA-A*02 population.
Capital raise requirements are still modest, based on the Company’s disciplined operations and low burn rate.
About FLAMINGO-01 Open Label Phase III Data
More than 1,500 patients have been screened at a screen rate of approximately 600-800 patients per year. The 250 patient non-HLA-A*02 arm is now fully enrolled, where all patients received GLSI-100, which is 5 times more treated patients and recurrence rate data than the approximately 50 patients treated in the Phase IIb trial. The Primary Immunization Series (PIS), which includes the first 6 GLSI-100 injections over the first 6 months and is required to reach peak protection, is followed by 5 booster injections given every 6 months to prolong the immune response, thereby providing longer-term protection.
In the non-HLA-A*02 arm, a preliminary analysis of recurrence rates after the PIS is completed shows an approximately 70-80% reduction in recurrence rate.
The non-HLA-A*02 arm is trending similarly to the Phase IIb trial results and hazard ratio where HLA-A*02 patients were treated and where breast cancer recurrences were reduced up to 80% compared to a 20-50% reduction in recurrence rate by other approved products.
The immune response at baseline prior to any GLSI-100 treatment, the increasing immune response during the PIS, and the safety profile of non-HLA-A*02 patients is trending similarly to the HLA-A*02 arms of FLAMINGO-01 and to the Phase IIb study.
The AACR Meeting 2026 delayed-type-hypersensitivity (DTH) poster and the ASCO Meeting 2026 injection site reaction (ISR) poster can be seen and downloaded at the bottom of the Phase III clinical trial tab on the Company’s website here.
As shown in both posters the frequency of DTH and ISR reactions increased statistically significantly over time.
As reported in Table 1 of each poster, each HLA-A type exhibited more frequent immune reactivity after treatment with GLSI-100 than at baseline.
Baseline DTH reaction prior to any treatment suggests that GP2 may be a natural antigen and that GP2 specific T cells may exist in some patients prior to any treatment with GLSI-100. Baseline immune response to GP2 prior to any vaccination with GP2 was also observed in the Phase IIb trial and is being observed in the blinded randomized arms of FLAMINGO-01, where HLA-A*02 only patients are being vaccinated.
Analysis of the open label data from FLAMINGO-01 has been conducted in a manner that maintains the study blind. The open label recurrence rate, immune response, and safety data is based on the patients enrolled to date in FLAMINGO-01 and the data provided by the clinical sites so far, which is not completed or fully reviewed, and is thus preliminary. While comparing any preliminary FLAMINGO-01 data to the Phase IIb clinical trial data may be possible, these preliminary results are not a prediction of future results, and the results at the end of the study may differ.
About GLSI-100 Phase IIb Study
In the prospective, randomized, single-blinded, placebo-controlled, multi-center (16 sites led by MD Anderson Cancer Center) Phase IIb clinical trial of HLA-A*02 breast cancer patients, 46 HER2/neu 3+ over-expressor patients were treated with GLSI-100, and 50 placebo patients were treated with GM-CSF alone. After 5 years of follow-up, there was an 80% or greater reduction in cancer recurrences in the HER2/neu 3+ patients who were treated with GLSI-100, followed, and remained disease free over the first 6 months, which we believe is the time required to reach peak immunity and thus maximum efficacy and protection. The Phase IIb posters and results can be summarized as follows and can be seen here:
80% or greater reduction in metastatic breast cancer recurrence rate over 5 years of follow-up with a peak immune response at 6 months and well-tolerated safety profile.
The PIS elicited a potent immune response as measured by local skin tests and immunological assays.
About FLAMINGO-01 and GLSI-100
FLAMINGO-01 (NCT05232916) is a Phase III clinical trial designed to evaluate the safety and efficacy of Fast Track designated GLSI-100 (GP2 + GM-CSF) in HER2 positive breast cancer patients who had residual disease or high-risk pathologic complete response at surgery and who have completed both neoadjuvant and postoperative adjuvant trastuzumab based treatment. The trial is led by Baylor College of Medicine and currently includes US and European clinical sites from university-based hospitals and academic and cooperative networks with plans to open up to 170-180 sites globally.
For more information on FLAMINGO-01, please visit the Company’s website here and clinicaltrials.gov here. Contact information and an interactive map of the majority of participating clinical sites can be viewed under the “Contacts and Locations” section. Please note that the interactive map is not viewable on mobile screens. Related questions and participation interest can be emailed to: [email protected]
About Breast Cancer and HER2/neu Positivity
One in eight U.S. women will develop invasive breast cancer over her lifetime. In the US and Europe, there are approximately 700,000 new breast cancer patients per year and 9.5 million breast cancer survivors. HER2 (human epidermal growth factor receptor 2) protein is a cell surface receptor protein that is expressed in a variety of common cancers, including in 75% of breast cancers at low (1+), intermediate (2+), and high (3+ or over-expressor) levels.
About Greenwich LifeSciences, Inc.
Greenwich LifeSciences is a clinical-stage biopharmaceutical company focused on the development of GP2, an immunotherapy to prevent breast cancer recurrences in patients who have previously undergone surgery. GP2 is a 9 amino acid transmembrane peptide of the HER2 protein, a cell surface receptor protein that is expressed in a variety of common cancers, including expression in 75% of breast cancers at low (1+), intermediate (2+), and high (3+ or over-expressor) levels. Greenwich LifeSciences has commenced a Phase III clinical trial, FLAMINGO-01. For more information on Greenwich LifeSciences, please visit the Company’s website at www.greenwichlifesciences.com and follow the Company’s Twitter at https://twitter.com/GreenwichLS.
Forward-Looking Statement Disclaimer
Statements in this press release contain “forward-looking statements” that are subject to substantial risks and uncertainties. All statements, other than statements of historical fact, contained in this press release are forward-looking statements. Forward-looking statements contained in this press release may be identified by the use of words such as “anticipate,” “believe,” “contemplate,” “could,” “estimate,” “expect,” “intend,” “seek,” “may,” “might,” “plan,” “potential,” “predict,” “project,” “target,” “aim,” “should,” “will,” “would,” or the negative of these words or other similar expressions, although not all forward-looking statements contain these words. Forward-looking statements are based on Greenwich LifeSciences Inc.’s current expectations and are subject to inherent uncertainties, risks and assumptions that are difficult to predict, including statements regarding the intended use of net proceeds from the public offering; consequently, actual results may differ materially from those expressed or implied by such forward-looking statements. Further, certain forward-looking statements are based on assumptions as to future events that may not prove to be accurate. These and other risks and uncertainties are described more fully in the section entitled “Risk Factors” in Greenwich LifeSciences’ Annual Report on the most recent Form 10-K for the year ended December 31, 2025, and other periodic reports filed with the Securities and Exchange Commission. Forward-looking statements contained in this announcement are made as of this date, and Greenwich LifeSciences, Inc. undertakes no duty to update such information except as required under applicable law.




















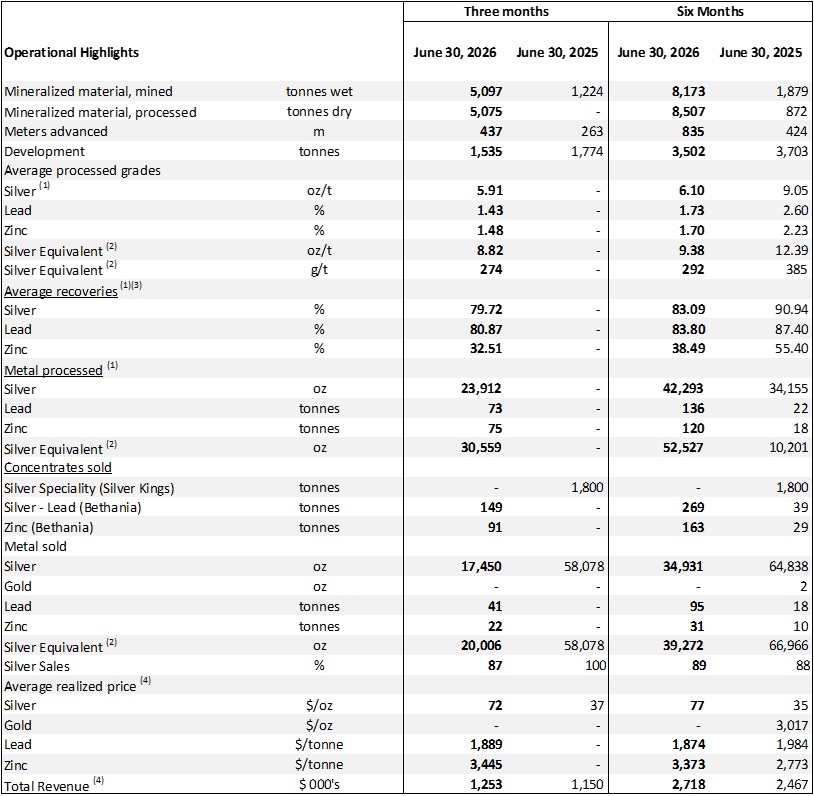{kind=link}