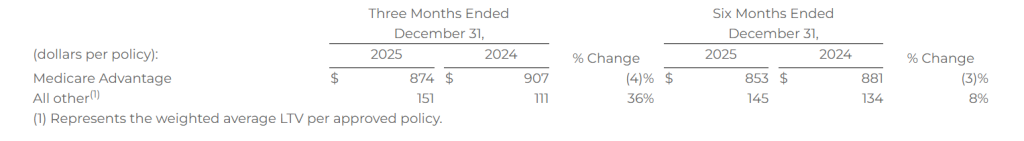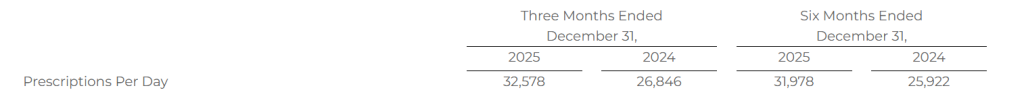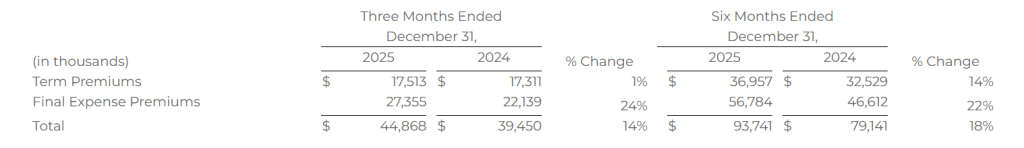Cocrystal Pharma, Inc. is a clinical-stage biotechnology company discovering and developing novel antiviral therapeutics that target the replication process of influenza viruses, coronaviruses (including SARS-CoV-2), hepatitis C viruses and noroviruses. Cocrystal employs unique structure-based technologies and Nobel Prize-winning expertise to create first- and best-in-class antiviral drugs. For further information about Cocrystal, please visit www.cocrystalpharma.com.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
CDI-988 Data Selected For Presentation At ICAR. Cocrystal announced that it has been selected to present data from its Phase 1 clinical trial and updates from the ongoing Phase 1b challenge study testing CDI-988 against norovirus infection at the 38th International Conference on Antiviral Research, to be held April 27 to May 1 in Prague, Czech Republic. We see the presentation at this important conference as recognition of the potential of CDI-988 for an indication that has serious medical and economic consequences.
Phase 1 and 1b Data Expected. We expect Dr. Sam Lee, President and Co-CEO, to present initial Phase 1 safety and tolerability data. Previously announced data from the single ascending dose (SAD) and multiple ascending dose (MAD) study showed safety and tolerability across all dose cohorts tested. Additional data from the ongoing Phase 1b norovirus challenge study testing CDI-988 as both a prophylactic and therapeutic may also be included.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Hims & Hers Health, Inc. has announced a definitive agreement to acquire Eucalyptus in a transaction valued at up to $1.15 billion, marking one of the most significant global expansion moves in the consumer telehealth sector to date. The deal positions Hims & Hers to accelerate its ambition of becoming the leading global consumer health platform, extending its reach well beyond the United States.
Under the terms of the agreement, approximately $240 million will be paid in cash at closing, with the remaining consideration structured as deferred payments and performance-based earnouts through early 2029. The company has emphasized that the transaction is designed to preserve balance sheet flexibility, with most of the funding expected to come from existing cash reserves and future U.S. operating cash flows. The acquisition is subject to regulatory approvals and is anticipated to close in mid-2026.
The strategic logic behind the transaction is straightforward: scale, infrastructure, and international expertise. Eucalyptus operates a portfolio of digital health brands across Australia, the United Kingdom, Germany, Japan, and Canada, and has served more than 775,000 customers. With an annual revenue run-rate exceeding $450 million and triple-digit year-over-year ARR growth throughout 2025, Eucalyptus brings both growth momentum and operational discipline to the combined platform.
For Hims & Hers, whose U.S. platform has built a reputation for direct-to-consumer access to personalized treatments across areas such as mental health, dermatology, sexual health, and weight management, the deal creates a ready-made international footprint. Rather than entering new markets from scratch, the company gains regulatory expertise, localized clinical infrastructure, and established consumer brands in key geographies.
Chief Executive Officer Andrew Dudum framed the acquisition as the next logical step in the company’s evolution, emphasizing that while healthcare challenges are universal, solutions must be tailored regionally. By integrating Eucalyptus’ local operating model with Hims & Hers’ technology platform and brand infrastructure, the company aims to expand access to personalized care globally while maintaining clinical quality and compliance standards.
Eucalyptus’ credibility adds weight to the expansion strategy. The company has facilitated nearly two million consultations and has published more than 20 peer-reviewed studies examining patient outcomes and adherence. It is also the first Australian telehealth provider accredited by the Australian Council on Healthcare Standards, underscoring its regulatory and clinical rigor.
Leadership continuity appears central to the integration plan. Eucalyptus CEO Tim Doyle will join Hims & Hers as Senior Vice President of International, overseeing global operations outside the U.S. His experience scaling digital healthcare businesses across multiple regulatory environments is expected to be instrumental as the company pushes deeper into Europe and Asia-Pacific markets.
From a competitive standpoint, the acquisition strengthens Hims & Hers’ position as pharmaceutical manufacturers, biotech firms, and diagnostic companies increasingly seek scalable digital distribution partners. The combined entity will offer capabilities ranging from online pharmacy fulfillment to concierge-style telehealth services, broadening its appeal across therapeutic categories.
If successfully executed, the deal could establish category leadership in Australia and meaningfully expand market share in the UK and Germany within the next two years. More broadly, it signals that consumer-centric digital health platforms are entering a new phase of consolidation and global ambition.
For investors and industry observers alike, this transaction is less about short-term expansion and more about building infrastructure for long-term dominance in global consumer healthcare.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
PrimeC Demonstrates Survival Benefit and 65% Mortality Risk Reduction. NeuroSense announced a Post-Phase 2b Analysis of its trial testing PrimeC in ALS. New data shows PrimeC patients had an additional 14 months (about 70%) survival with 65% reduction in risk of death. These improvements in overall survival correlate with previous Phase 2b Paradigm data that showed improvements in several endpoints of function, biomarkers, and survival.
New Data Shows Continued Improvement In Survival. The newly released data show the PrimeC treated patients had a median survival benefit of 36.3 months compared with 21.4 months for the group that received placebo then PrimeC during the extension study. This improvement of about 14.9 months was a benefit of 70% in survival. The Hazard Ratio (HR, the probability of an event occurring) reduced risk of death by 65% (p=0.0037).
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Weight-loss drugs have moved from niche medical treatments to mainstream consumer products. Television commercials, celebrity endorsements, and telehealth platforms have helped propel GLP-1–based therapies into the public consciousness — and into millions of medicine cabinets.
But as demand has surged, so have tensions across the healthcare, regulatory, and investment landscape.
That tension came sharply into focus today after Hims & Hers Health, Inc. (HIMS) shares fell more than 20% following news that Novo Nordisk has filed a lawsuit seeking to permanently block Hims from selling compounded versions of drugs that allegedly infringe on Novo’s patents — including versions tied to Wegovy, its blockbuster obesity treatment.
The dispute highlights a broader reckoning underway in the fast-growing — and fast-changing — obesity drug market.
A Market Built on Demand — and a Regulatory Loophole
GLP-1 drugs such as Wegovy (Novo Nordisk) and Zepbound/Mounjaro (Eli Lilly) have reshaped expectations around medical weight loss. Unprecedented demand led analysts to project a global obesity drug market of $150 billion to $200 billion by the early 2030s.
But demand quickly ran into supply constraints, high prices, and limited insurance coverage. That gap created an opening for compounded versions of GLP-1 drugs — products mixed by pharmacies and prescribed on a case-by-case basis under the Federal Food, Drug, and Cosmetic Act.
Under U.S. law, compounding is permitted in limited circumstances, such as:
When a patient cannot tolerate an ingredient in a branded drug
When a specific dosage or formulation is medically necessary
When an FDA-approved drug is in short supply
Novo has estimated that as many as 1.5 million Americans are currently using compounded GLP-1 drugs.
Telehealth companies like Hims moved aggressively into this space, marketing lower-cost alternatives to branded therapies — often directly to cash-pay consumers.
Novo vs. Hims: From Tension to Litigation
Novo Nordisk’s lawsuit represents a major escalation.
The company is asking the court to:
Permanently ban Hims from selling compounded versions of its drugs
Recover damages for alleged patent infringement
Novo argues that Hims’ compounded products contain semaglutide, the active ingredient in Wegovy, which is protected by U.S. patents through 2032. Importantly, Novo has stated that semaglutide is no longer in short supply in the U.S. — undermining one of the key legal justifications for compounding.
Hims, for its part, has argued that its products are legal because they are “personalized” in dosage. The company had planned to offer an oral obesity pill for as little as $49 for the first month, roughly $100 less than Novo’s approved Wegovy pill.
However, the pressure intensified last week when:
Hims said it would stop offering its newly launched obesity pill copycat
The FDA announced it planned to take legal action against Hims
Federal regulators said they would restrict access to GLP-1 ingredients used in non-approved compounded drugs
The FDA indicated it may refer the matter to the Department of Justice over potential violations of federal law
In a public statement, Hims called Novo’s lawsuit “a blatant attack by a Danish company on millions of Americans who rely on compounded medications for access to personalized care,” accusing Big Pharma of weaponizing the U.S. judicial system to limit consumer choice.
FDA Scrutiny Raises the Stakes
The FDA has made clear it is increasingly concerned about the quality, safety, and legality of compounded GLP-1 products.
Unlike branded drugs:
Compounded drugs are not FDA-approved
They have not undergone clinical trials to demonstrate efficacy
Oversight is more fragmented
According to legal experts, potential FDA enforcement actions could include:
Warning letters
Court injunctions (with DOJ involvement)
Administrative seizure of products
Novo and Eli Lilly have both taken aggressive steps over the past two years to crack down on compounding pharmacies and marketers. Novo has reportedly filed around 130 lawsuits related to deceptive marketing practices and consumer fraud, while Lilly has pursued similar actions tied to tirzepatide, its active ingredient.
Investors Reassess the Obesity Drug Opportunity
Beyond the immediate legal headlines, the episode underscores a broader shift in how Wall Street views the obesity drug market.
While demand remains strong, expectations around pricing power and long-term market size are being recalibrated:
Forecasts for the global obesity market have fallen roughly 30%, to around $100 billion by 2030
The once-common $150 billion target has been pushed out to 2035 by some analysts
Jefferies recently cut its peak market estimate by 20%, projecting a peak of $80 billion
As Jefferies analyst Michael Leuchten put it: “That $150 billion pie is gone, even if you’re very bullish on volumes.”
Competition is intensifying as well. Novo and Lilly remain the dominant players, but falling U.S. prices, the expected entry of new drugs, and eventual generic competition are reshaping the outlook — particularly in the cash-pay consumer segment.
What This Means Going Forward
For consumers, the crackdown on compounding could limit access to lower-cost alternatives — at least in the near term.
For telehealth companies, the legal and regulatory risks around drug development and distribution are becoming harder to ignore.
And for investors, the GLP-1 market is entering a new phase: one where growth remains substantial, but margins, market share, and timelines are far less certain than they appeared just a year ago.
The obesity drug boom is real. But as the fight between Novo Nordisk, Hims, the FDA, and regulators shows, the path forward will be shaped as much by courts and policymakers as by science and demand.
Patrick McCann, CFA, Research Analyst, Noble Capital Markets, Inc.
Michael Kupinski, Director of Research, Equity Research Analyst, Digital, Media & Technology , Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Fiscal Q2 results. SelectQuote reported fiscal Q2 revenue of $537.1 million, above our $520.0 million estimate, driven by stronger-than-expected Senior performance. Adj. EBITDA of $84.7 million exceeded our $82.0 million forecast, reflecting near-record 39% adj. EBITDA margins in Senior that more than offset pharmacy reimbursement pressure.
Medicare Advantage headwinds. Management cited pressure from a large national carrier’s decision to reduce strategic marketing spend across all channels. We believe this reflects a deliberate effort to moderate enrollment growth and protect plan profitability following above-trend member additions, rather than any deterioration in underlying demand.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Second Quarter of Fiscal Year 2026 – Consolidated Earnings Highlights
Revenue of $537.1 million
Net income of $69.3 million
Adjusted EBITDA* of $84.7 million
Fiscal Year 2026 Guidance Ranges:
Revenue expected in a range of $1.61 billion to $1.71 billion
Adjusted EBITDA* expected in a range of $90 million to $100 million
Second Quarter Fiscal Year 2026 – Segment Highlights
Senior
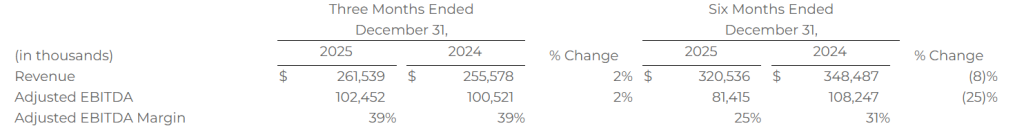
Revenue of $261.5 million
Adjusted EBITDA of $102.5 million
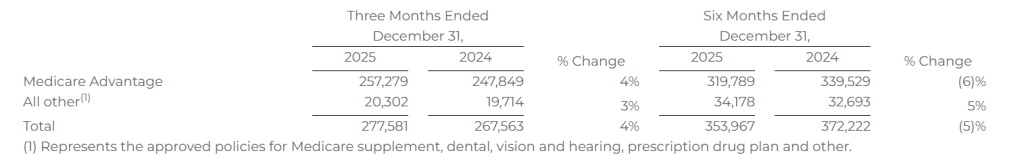
Approved Medicare Advantage policies of 257,279
Healthcare Services
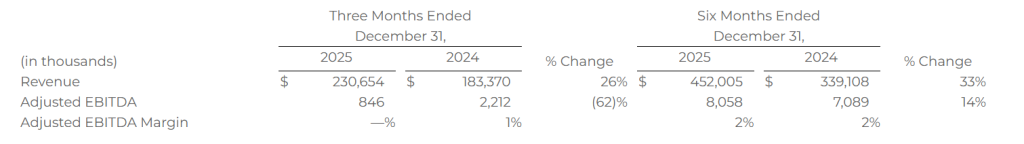
Revenue of $230.7 million
Adjusted EBITDA of $0.8 million
113,483 SelectRx members
Life
Revenue of $43.6 million
Adjusted EBITDA of $5.6 million
OVERLAND PARK, Kan.–(BUSINESS WIRE)– SelectQuote, Inc. (NYSE: SLQT) reported consolidated revenue for the second quarter of fiscal year 2026 of $537.1 million compared to consolidated revenue for the second quarter of fiscal year 2025 of $481.1 million. Consolidated net income for the second quarter of fiscal year 2026 was $69.3 million compared to consolidated net income for the second quarter of fiscal year 2025 of $53.2 million. Finally, consolidated Adjusted EBITDA* for the second quarter of fiscal year 2026 was $84.7 million compared to consolidated Adjusted EBITDA* for the second quarter of fiscal year 2025 of $87.5 million.
Tim Danker, SelectQuote Chief Executive Officer, “This year’s AEP again highlighted the strength and consistency of SelectQuote’s operating model. Despite continued volatility in Medicare Advantage benefit structures, our team delivered another season of high‑quality execution, with strong agent productivity and marketing efficiency driving 39% Adjusted EBITDA* margins for our Senior business. At the same time, our rapidly growing Healthcare Services segment, led by SelectRx, continues to provide meaningful clinical value for members and attractive long‑term economics for our platform. The combination of improved medication adherence, lower waste, and better patient outcomes reinforces SelectRx as an increasingly important driver of value creation for the company and broader pharmacy ecosystem.
Our revised fiscal 2026 guidance reflects two discrete, partner‑driven headwinds: a national carrier’s decision to constrain additional MA policy volume by curtailing strategic marketing spend across all channels, and the previously communicated PBM reimbursement changes. Neither impact related to our internal performance, which remained strong. While these developments are frustrating, they do not alter our conviction in the long‑term earnings power of SelectQuote’s comprehensive healthcare platform.
What continues to give us confidence is the consistency of our underlying operational execution. Regardless of the market backdrop, our teams have demonstrated the ability to drive efficiency, deliver value for partners and beneficiaries, and maintain strong margin discipline. Coupled with our improved balance sheet flexibility, we believe this operational consistency positions SelectQuote to deliver meaningful cash‑flow generation for shareholders in the quarters and years ahead.”
* See “Non-GAAP Financial Measures” below.
Segment Results
We currently have three reportable segments: 1) Senior, 2) Healthcare Services and 3) Life. The performance measures of the segments include total revenue and adjusted EBITDA. Costs of commissions and other services revenue, cost of goods sold-pharmacy revenue, marketing and advertising, selling, general, and administrative, and technical development operating expenses that are directly attributable to a segment are reported within the applicable segment. Indirect costs of revenue, marketing and advertising, selling, general, and administrative, and technical development operating expenses are allocated to each segment based on varying metrics such as headcount.
Senior
Financial Results
The following table provides the financial results for the Senior segment for the periods presented:
Operating Metrics
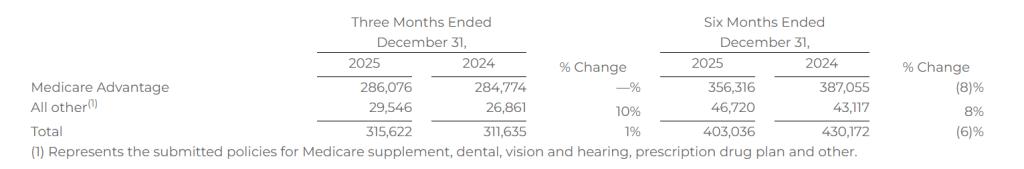
Submitted Policies
Submitted policies are counted when an individual completes an application with our licensed agent and provides authorization to the agent to submit the application to the insurance carrier partner. The applicant may have additional actions to take before the application will be reviewed by the insurance carrier.
The following table shows the number of submitted policies for the periods presented:
Approved Policies
Approved policies represents the number of submitted policies that were approved by our insurance carrier partners for the identified product during the indicated period. Not all approved policies will go in force.
The following table shows the number of approved policies for the periods presented:
Lifetime Value of Commissions per Approved Policy
Lifetime value of commissions per approved policy represents commissions estimated to be collected over the estimated life of an approved policy based on multiple factors, including but not limited to, contracted commission rates, carrier mix and expected policy persistency with applied constraints. The lifetime value of commissions per approved policy is equal to the sum of the commission revenue due upon the initial sale of a policy, and when applicable, an estimate of future renewal commissions.
The following table shows the lifetime value of commissions per approved policy for the periods presented:
Healthcare Services
Financial Results
The following table provides the financial results for the Healthcare Services segment for the periods presented:
Operating Metrics
Members
The total number of SelectRx members represents the amount of active customers to which an order has been shipped and the prescriptions per day represents the total average prescriptions shipped per business day. These two metrics are the primary drivers of revenue for Healthcare Services.
The following table shows the total number of SelectRx members as of the periods presented:
The total number of SelectRx members increased by 17% as of December 31, 2025, compared to December 31, 2024, due to our strategy to grow SelectRx membership.
The following table shows the average prescriptions shipped per day for the periods presented:
Combined Senior and Healthcare Services – Consumer Per Unit Economics
Combined Senior and Healthcare Services consumer per unit economics represents total MA and MS commissions; other product commissions; other revenues, including revenues from Healthcare Services; and operating expenses associated with Senior and Healthcare Services, each shown per number of approved MA and MS policies over a given time period. Management assesses the business on a per-unit basis to help ensure that the revenue opportunity associated with a successful policy sale is attractive relative to the marketing acquisition cost. Because not all acquired leads result in a successful policy sale, all per-policy metrics are based on approved policies, which is the measure that triggers revenue recognition.
The MA and MS commission per MA/MS policy represents the LTV for policies sold in the period. Other commission per MA/MS policy represents the LTV for other products sold in the period, including DVH prescription drug plan, and other products, which management views as additional commission revenue on our agents’ core function of MA/MS policy sales. Pharmacy revenue per MA/MS policy represents revenue from SelectRx, and other revenue per MA/MS policy represents revenue from Population Health, production bonuses, marketing development funds, lead generation revenue, and adjustments from the Company’s reassessment of its cohorts’ transaction prices. Total operating expenses per MA/MS policy represents all of the operating expenses within Senior and Healthcare Services. The revenue to customer acquisition cost (“CAC”) multiple represents total revenue as a multiple of total marketing acquisition cost, which represents the direct costs of acquiring leads. These costs are included in marketing and advertising expense within the total operating expenses per MA/MS policy.
The following table shows combined Senior and Healthcare Services consumer per unit economics for the periods presented. Based on the seasonality of Senior and the fluctuations between quarters, we believe that the most relevant view of per unit economics is on a rolling 12-month basis. All per MA/MS policy metrics below are based on the sum of approved MA/MS policies, as both products have similar commission profiles.
Total revenue per MA/MS policy increased 23% for the twelve months ended December 31, 2025, compared to the twelve months ended December 31, 2024, primarily due to the increase in pharmacy revenue. Total operating expenses per MA/MS policy increased 31% for the twelve months ended December 31, 2025, compared to the twelve months ended December 31, 2024, driven by an increase in cost of goods sold-pharmacy revenue for Healthcare Services due to the growth of the business.
Life
Financial Results
The following table provides the financial results for the Life segment for the periods presented:
Operating Metrics
Life premium represents the total premium value for all policies that were approved by the relevant insurance carrier partner and for which the policy document was sent to the policyholder and payment information was received by the relevant insurance carrier partner during the indicated period. Because our commissions are earned based on a percentage of total premium, total premium volume for a given period is the key driver of revenue for our Life segment.
The following table shows term and final expense premiums for the periods presented:
Earnings Conference Call
SelectQuote, Inc. will host a conference call with the investment community on February 5, 2025 beginning at 8:00 a.m. ET. To register for this conference call, please use this link: https://events.q4inc.com/analyst/199368355?pwd=c0a3KINj. After registering, a confirmation will be sent via email, including dial-in details and unique conference call codes for entry. Registration is open through the live call, but to ensure you are connected for the full call we suggest registering at least 10 minutes before the start of the call. The event will also be webcasted live via our investor relations website https://ir.selectquote.com/investor-home/default.aspx.
Non-GAAP Financial Measures
This release includes certain non-GAAP financial measures intended to supplement, not substitute for, comparable GAAP measures. To supplement our financial statements presented in accordance with GAAP and to provide investors with additional information regarding our GAAP financial results, we have presented in this release Adjusted EBITDA, which, when presented on a consolidated basis, is a non-GAAP financial measure. This non-GAAP financial measure is not based on any standardized methodology prescribed by GAAP and is not necessarily comparable to any similarly titled measure presented by other companies. We define Adjusted EBITDA as net income (loss) plus interest expense, income taxes, depreciation and amortization, changes in fair value of warrant liabilities, and certain add-backs for non-cash or non-recurring expenses, including restructuring and share-based compensation expenses. The most directly comparable GAAP measure is net income (loss). We monitor and have presented in this release Adjusted EBITDA because it is a key measure used by our management and Board of Directors to understand and evaluate our operating performance, establish budgets, and develop operational goals for managing our business. In particular, we believe that excluding the impact of these expenses in calculating Adjusted EBITDA can provide a useful measure for period-to-period comparisons of our core operating performance.
A reconciliation of the differences between Adjusted EBITDA and its most directly comparable GAAP measure, net income (loss), is presented below on page 15. The Company is unable to provide a quantitative reconciliation of forward-looking Adjusted EBITDA to its most directly comparable GAAP measure without unreasonable effort because it is not possible to predict certain information included in the calculation of such GAAP measure, including the fair value of outstanding warrants to purchase shares of the Company’s common stock. The unavailable information could have a significant impact on the Company’s GAAP financial results.
Forward Looking Statements
This release contains forward-looking statements. These forward-looking statements reflect our current views with respect to, among other things, future events and our financial performance. These statements are often, but not always, made through the use of words or phrases such as “may,” “should,” “could,” “predict,” “potential,” “believe,” “will likely result,” “expect,” “continue,” “will,” “anticipate,” “seek,” “estimate,” “intend,” “plan,” “projection,” “would” and “outlook,” or the negative version of those words or other comparable words or phrases of a future or forward-looking nature. These forward-looking statements are not historical facts, and are based on current expectations, estimates and projections about our industry, management’s beliefs and certain assumptions made by management, many of which, by their nature, are inherently uncertain and beyond our control. Accordingly, we caution you that any such forward-looking statements are not guarantees of future performance and are subject to risks, assumptions and uncertainties that are difficult to predict. Although we believe that the expectations reflected in these forward-looking statements are reasonable as of the date made, actual results may prove to be materially different from the results expressed or implied by the forward-looking statements. There are or will be important factors that could cause our actual results to differ materially from those indicated in these forward-looking statements, including, but not limited to, the following: our reliance on a limited number of insurance carrier partners and any potential termination of those relationships or failure to develop new relationships; existing and future laws and regulations affecting the health insurance market; changes in health insurance products offered by our insurance carrier partners and the health insurance market generally; insurance carriers offering products and services directly to consumers; changes to commissions paid by insurance carriers and underwriting practices; competition with brokers, exclusively online brokers and carriers who opt to sell policies directly to consumers; competition from government-run health insurance exchanges; developments in the U.S. health insurance system; our dependence on revenue from carriers in our senior segment and downturns in the senior health as well as life, automotive and home insurance industries; our ability to develop new offerings and penetrate new vertical markets; risks from third-party products; failure to enroll individuals during the Medicare annual enrollment period; our ability to attract, integrate and retain qualified personnel; our dependence on lead providers and ability to compete for leads; failure to obtain and/or convert sales leads to actual sales of insurance policies; access to data from consumers and insurance carriers; accuracy of information provided from and to consumers during the insurance shopping process; cost-effective advertisement through internet search engines; ability to contact consumers and market products by telephone; global economic conditions, including inflation; disruption to operations as a result of future acquisitions; significant estimates and assumptions in the preparation of our financial statements; impairment of goodwill; potential litigation and other legal proceedings or inquiries; our existing and future indebtedness; our ability to maintain compliance with our debt covenants; access to additional capital; failure to protect our intellectual property and our brand; fluctuations in our financial results caused by seasonality; accuracy and timeliness of commissions reports from insurance carriers; timing of insurance carriers’ approval and payment practices; factors that impact our estimate of the constrained lifetime value of commissions per policyholder; changes in accounting rules, tax legislation and other legislation; disruptions or failures of our technological infrastructure and platform; failure to maintain relationships with third-party service providers; cybersecurity breaches or other attacks involving our systems or those of our insurance carrier partners or third-party service providers; our ability to protect consumer information and other data; failure to market and sell Medicare plans effectively or in compliance with laws; and other factors related to our pharmacy business, including manufacturing or supply chain disruptions, access to and demand for prescription drugs, changes in reimbursement rates under our contracts with pharmacy benefit managers, and regulatory changes or other industry developments that may affect our pharmacy operations. For a further discussion of these and other risk factors that could impact our future results and performance, see the section entitled “Risk Factors” in the most recent Annual Report on Form 10-K (the “Annual Report”) and subsequent periodic reports filed by us with the Securities and Exchange Commission. Accordingly, you should not place undue reliance on any such forward-looking statements. Any forward-looking statement speaks only as of the date on which it is made, and, except as otherwise required by law, we do not undertake any obligation to publicly update or review any forward-looking statement, whether as a result of new information, future developments or otherwise.
About SelectQuote:
Founded in 1985, SelectQuote (NYSE: SLQT) pioneered the model of providing unbiased comparisons from multiple, highly-rated insurance companies, allowing consumers to choose the policy and terms that best meet their unique needs. Two foundational pillars underpin SelectQuote’s success: a strong force of highly-trained and skilled agents who provide a consultative needs analysis for every consumer, and proprietary technology that sources and routes high-quality leads. Today, the Company operates an ecosystem offering high touchpoints for consumers across insurance, pharmacy, and virtual care.
With an ecosystem offering engagement points for consumers across insurance, Medicare, pharmacy, and value-based care, the company now has three core business lines: SelectQuote Senior, SelectQuote Healthcare Services, and SelectQuote Life. SelectQuote Senior serves the needs of a demographic that sees around 10,000 people turn 65 each day with a range of Medicare Advantage and Medicare Supplement plans. SelectQuote Healthcare Services is comprised of the SelectRx Pharmacy, a Patient-Centered Pharmacy Home™ (PCPH) accredited pharmacy, SelectPatient Management, a provider of chronic care management services, and Healthcare Select which proactively connects consumers with a wide breadth of healthcare services supporting their needs.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Phase 1b Data For Second Year After Transplantation Presented. Eledon presented data from its Phase 1b trial at the American Society of Transplant Surgeons (ASTS) meeting in January 2026. The presentation included data from 8 patients that had reached 24 months after transplantation, compared with 12 patients evaluated 12 months after transplantation presented in August 2025. These new data show a continued improvement in kidney function during the second year.
New Data Show Durability With Improvements. The 24-month data shows eGFR in tegoprubart patients continued to improve during months 12 to 24 after transplantation. The eGFR levels were restored to normal levels within 1 month after transplantation and were maintained for up to 2 years. Although this is a small number of patients, we see the result as consistent with prior data and our expectations for organ survival.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Unicycive Announced FDA Acceptance Of The NDA. Unicycive announced FDA acceptance of its resubmission of the New Drug Application (NDA) for OLC (oxylanthanum citrate). The resubmitted application has been classified as a Class II complete response, with a six-month review period. June 29, 2026 is the new PDUFA date, the statutory date for the application to be answered. This is consistent with our expected timeframe for OLC approval and launch.
We See NDA Acceptance As A Significant Milestone. In June 2025, an FDA manufacturing inspection found compliance deficiencies at the facility of a contract manufacturer. This stopped the NDA approval process just weeks before the PDUFA (Prescription Drug User Fee Act) date of June 28, 2025. The review of the preclinical, clinical, safety, and manufacturing data had been completed. We believe this will result in prompt approval.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Cardiff Made Two Significant Announcements. New data from the Phase 2 CRDF-004 trial testing onvansertib as a first line treatment for metastatic colorectal cancer was announced as expected. Patients in the high-dose onvansertib group showed a large benefit in overall response rates (ORR) and progression free survival (PFS). Separately, the CEO and CFO have left the company. Board Member Dr. Mani Mohindru was named Interim CEO.
Phase 2 Trial Design. As discussed in our January 5 report, CDRF-004 is a Phase 2 dose-finding trial testing two doses of onvansertib in combination with two standard-of-care (SOC) regimens against the standard of care regimens alone. It enrolled 110 patients with RAS-mutated metastatic colorectal cancer, mCRC. Its primary endpoint is objective response rate (ORR). Secondary endpoints include progression-free survival (PFS), duration of response (DOR) and safety. These endpoints were selected to guide the design of Phase 3.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Data from eight participants continue to support safety and tolerability profile of tegoprubart
Mean eGFR increased over the measurement period, from 67.0 mL/min/1.73 m² at 12 months to 74.2 mL/min/1.73 m² at 24 months
IRVINE, Calif., Jan. 23, 2026 (GLOBE NEWSWIRE) — Eledon Pharmaceuticals, Inc. (“Eledon”) (Nasdaq: ELDN) today announced that it will present 24-month follow-up data from eight patients enrolled in the Phase 1b trial long-term extension evaluating tegoprubart in kidney transplantation at the American Society of Transplant Surgeons Winter Symposium, taking place January 23–25, 2026, in Scottsdale, Arizona.
There were no episodes of biopsy-proven acute rejection, graft loss, death, new-onset diabetes mellitus, or de novo donor-specific antibody formation during the study period. Mean estimated glomerular filtration rate (eGFR) increased over the measurement period, from 67.0 mL/min/1.73 m² at 12 months to 74.2 mL/min/1.73 m² at 24 months.
Details on the poster presentation are below:
Title: Long-Term Outcomes of a Phase 1, Single Arm Cohort of De Novo Kidney Transplant Recipients Treated with Tegoprubart, an Anti-CD40L Antibody, as the Core Immunosuppression Regimen Poster: #62 Session Title: Poster Session B Date: Friday, January 23, 2026, from 5:45 – 7:15 p.m. PT
Eledon Pharmaceuticals, Inc. is a clinical stage biotechnology company that is developing immune-modulating therapies for the management and treatment of life-threatening conditions. The Company’s lead investigational product is tegoprubart, an anti-CD40L antibody with high affinity for the CD40 Ligand, a well-validated biological target that has broad therapeutic potential. The central role of CD40L signaling in both adaptive and innate immune cell activation and function positions it as an attractive target for non-lymphocyte depleting, immunomodulatory therapeutic intervention. The Company is building upon a deep historical knowledge of anti-CD40 Ligand biology to conduct preclinical and clinical studies in kidney allograft transplantation, xenotransplantation, islet cell transplantation, and amyotrophic lateral sclerosis (ALS). Eledon is headquartered in Irvine, California. For more information, please visit the Company’s website at www.eledon.com.
Follow Eledon Pharmaceuticals on social media: LinkedIn; Twitter
Forward-Looking Statements
This press release contains forward-looking statements that involve substantial risks and uncertainties. Any statements about the company’s future expectations, plans and prospects, including statements about planned clinical trials, the development of product candidates, as well as other statements containing the words “believes,” “anticipates,” “plans,” “expects,” “estimates,” “intends,” “predicts,” “projects,” “targets,” “looks forward,” “could,” “may,” and similar expressions, constitute forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements are inherently uncertain and are subject to numerous risks and uncertainties, including: our short operating history and shifts in our business strategy; our operating losses since inception; our need for additional funding to develop our lead drug candidate and our ability to secure additional funding on acceptable terms or at all; the impact of issuances of our common stock, including in the possibility of dilution or a decline in our stock price; our ability to successfully develop our product candidates; unfavorable global economic and financial market conditions; the regulatory environment of our business and our ability to obtain required regulatory approvals; results of non-clinical studies and clinical trials, and risks that non-clinical studies or early clinical trials may not be predictive of results of later-stage clinical trials; delays or difficulties in enrollment of patients in clinical trials; our ability to attract and retain our executives and key employees; legislation of the pharmaceutical and healthcare industries; cybersecurity and data privacy risks; the ability of our products to achieve marketing approval; competition in our industry; our ability to obtain insurance coverage; our dependence on contract research organizations; our ability to protect our intellectual property; public health crises; our ability to establish and maintain proper and effective internal control over financial reporting and other risks disclosed in our Quarterly Report on Form 10-Q for the quarter ended September 30, 2025, filed with the Securities and Exchange Commission on November 14, 2025. Actual results may differ materially from those indicated by such forward-looking statements as a result of various factors. These risks and uncertainties, as well as other risks and uncertainties that could cause the company’s actual results to differ materially from the forward-looking statements contained herein, are discussed in our quarterly 10-Q, annual 10-K, and other filings with the U.S. Securities and Exchange Commission, which can be found at www.sec.gov. Any forward-looking statements contained in this press release speak only as of the date hereof and not of any future date, and the company expressly disclaims any intent to update any forward-looking statements, whether as a result of new information, future events or otherwise.
MALVERN, Pa., Jan. 23, 2026 (GLOBE NEWSWIRE) — Ocugen, Inc. (Nasdaq: OCGN), a pioneering biotechnology leader in gene therapies for blindness diseases, today announced the closing of its previously announced underwritten registered direct offering of 15,000,000 shares of its common stock at an offering price of $1.50 per share of common stock for net proceeds of $20.85 million, after deducting commissions and other estimated offering expenses payable by Ocugen. The financing was led by RTW Investments, with additional participation from new and existing investors.
Ocugen intends to use the net proceeds from the offering for general corporate purposes, capital expenditures, working capital, and general and administrative expenses and anticipates that the net proceeds will extend the company’s cash runway into the fourth quarter of 2026.
Oppenheimer & Co. acted as the sole book-running manager for the offering.
The offering was made pursuant to a shelf registration statement on Form S-3 (File No. 333-278774) previously filed with the Securities and Exchange Commission (the “SEC”) on April 18, 2024, which became effective on May 1, 2024. The offering was made only by means of a prospectus and prospectus supplement that form a part of the registration statement. A prospectus supplement relating to and describing the terms of the offering has been filed with the SEC. Copies of the prospectus supplement and the accompanying base prospectus relating to the offering, may be obtained by visiting the SEC’s website at www.sec.gov or by contacting Oppenheimer & Co. Inc. Attention: Syndicate Prospectus Department, 85 Broad Street, 26th Floor, New York, NY 10004, or by telephone at (212) 667-8055, or by email at EquityProspectus@opco.com.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of, these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of such state or jurisdiction.
AboutOcugen,Inc.
Ocugen, Inc. is a pioneering biotechnology leader in gene therapies for blindness diseases. Our breakthrough modifier gene therapy platform has the potential to address significant unmet medical need for large patient populations through our gene-agnostic approach. Unlike traditional gene therapies and gene editing, Ocugen’s modifier gene therapies address the entire disease—complex diseases that are potentially caused by imbalances in multiple gene networks. Currently we have programs in development for inherited retinal diseases and blindness diseases affecting millions across the globe, including retinitis pigmentosa, Stargardt disease, and geographic atrophy—late stage dry age-related macular degeneration.
This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995, which are subject to risks and uncertainties. Such forward-looking statements within this press release include, without limitation, statements regarding Ocugen’s expectations regarding the anticipated use of proceeds. We may, in some cases, use terms such as “predicts,” “believes,” “potential,” “proposed,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Such statements are subject to numerous important factors, risks and uncertainties that may cause actual events or results to differ materially from our current expectations, such as market and other conditions. Further, certain forward-looking statements are based on assumptions as to future events that may not prove to be accurate, including the Company’s expected cash runway and various other factors. These and other risks and uncertainties are more fully described in our periodic filings with the SEC, including the risk factors described in the section entitled “Risk Factors” in the quarterly and annual reports that we file with the SEC. Any forward-looking statements that we make in this press release speak only as of the date of this press release. Except as required by applicable law, we assume no obligation to update forward-looking statements contained in this press release whether as a result of new information, future events, changed circumstances or otherwise, after the date of this press release.
OcugenContact:
Tiffany Hamilton AVP, Head of Communications Tiffany.Hamilton@Ocugen.com
OVERLAND PARK, Kan.–(BUSINESS WIRE)– SelectQuote, Inc. (NYSE: SLQT), a leading distributor of Medicare insurance policies and owner of a rapidly growing Healthcare Services platform, today announced it will release its fiscal second quarter 2026 financial results before market open on Thursday, February 5, 2026. Chief Executive Officer, Tim Danker, and Chief Financial Officer, Ryan Clement, will host a conference call on the day of the release (February 5, 2026) at 8:00 am ET to discuss the results.
For those interested in dialing into the conference call, please register using this link. After registering, a confirmation will be sent via email, including dial in details and unique conference call codes for entry. Registration is open through the live call, but to ensure you are connected for the full call, we suggest registering a day in advance or at least 10 minutes before the start of the call.
About SelectQuote:
Founded in 1985, SelectQuote (NYSE: SLQT) pioneered the model of providing unbiased comparisons from multiple, highly rated insurance companies, allowing consumers to choose the policy and terms that best meet their unique needs. Two foundational pillars underpin SelectQuote’s success: a strong force of highly trained and skilled agents who provide a consultative needs analysis for every consumer, and proprietary technology that sources and routes high-quality leads. Today, the Company operates an ecosystem offering high touchpoints for consumers across insurance, pharmacy, and virtual care.
With an ecosystem offering engagement points for consumers across insurance, Medicare, pharmacy, and value-based care, the company now has three core business lines: SelectQuote Senior, SelectQuote Healthcare Services, and SelectQuote Life. SelectQuote Senior serves the needs of a demographic that sees around 10,000 people turn 65 each day with a range of Medicare Advantage and Medicare Supplement plans. SelectQuote Healthcare Services is comprised of the SelectRx Pharmacy, a Patient-Centered Pharmacy Home™ (PCPH) accredited pharmacy, SelectPatient Management, a provider of chronic care management services, and Healthcare Select, which proactively connects consumers with a wide breadth of healthcare services supporting their needs.
GSK’s agreement to acquire RAPT Therapeutics for $58 per share in cash underscores a growing trend in biotech investing: large pharmaceutical companies are increasingly turning to small-cap innovators to fill critical gaps in their pipelines. For small-cap investors, the deal offers a clear example of how differentiated science, even at the clinical-stage level, can command a meaningful premium.
Under the terms of the agreement, GSK will acquire RAPT Therapeutics for an estimated equity value of $2.2 billion, or approximately $1.9 billion net of cash acquired. The transaction is expected to close in the first quarter of 2026, pending customary regulatory approvals and shareholder tender conditions. Shares of RAPT surged following the announcement, reflecting both the attractive takeover premium and validation of the company’s lead asset.
At the center of the deal is ozureprubart, a long-acting anti-immunoglobulin E (IgE) monoclonal antibody currently in Phase IIb development for prophylactic protection against food allergens. IgE is a clinically validated target and is responsible for roughly 94% of severe food allergy reactions, making it one of the most established mechanisms in allergy treatment. However, existing anti-IgE therapies require injections every two to four weeks, creating a significant burden for patients—most of whom are children.
Ozureprubart’s potential differentiator lies in its dosing profile. The therapy is designed to be administered once every 12 weeks, which could dramatically improve patient compliance and expand treatment eligibility to an estimated 25% of patients who are currently unable to use standard therapies. If successful in late-stage trials, ozureprubart could represent a best-in-class option in a market with substantial unmet medical need.
From GSK’s perspective, the acquisition strengthens its Respiratory, Immunology, and Inflammation pipeline and leverages its existing commercial footprint in allergy and immunology. For a company of GSK’s scale, the upfront investment is manageable, while the long-term upside could be significant. In the U.S. alone, more than 17 million people are diagnosed with food allergies, with over 1.3 million experiencing severe reactions that often require emergency care.
For small-cap investors, the RAPT deal is instructive. RAPT was a clinical-stage company without an approved product, yet it attracted a multibillion-dollar buyout based on a single, well-positioned asset targeting a validated pathway. This reinforces the idea that big pharma is willing to pay for de-risked science, especially when it addresses large, underserved markets and fits cleanly into an existing commercial infrastructure.
The transaction also highlights the importance of platform credibility. RAPT’s focus on immunology and its ability to advance ozureprubart into mid-stage clinical development made it a credible acquisition target rather than a speculative bet.
While not every small-cap biotech will see a similar outcome, GSK’s acquisition of RAPT Therapeutics serves as a reminder that disciplined execution, clear differentiation, and alignment with big pharma priorities can create substantial shareholder value—even before commercialization.