Research News and Market Data on TNXP
June 25, 2024 8:00am EDT
Tonix’s live virus vaccine TNX-801 is designed to provide long-term protection from mpox and smallpox with one dose
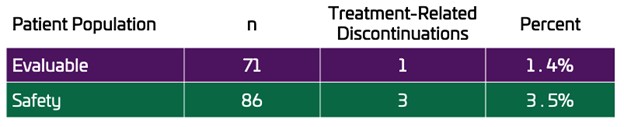
TNX-801 vaccination demonstrated efficacy in protecting animals from lethal challenge with intratracheal monkeypox
Clade II mpox is now endemic in the U.S. with >30,000 cases reported since May 20221 and Clade I mpox is endemic in the Democratic Republic of the Congo2
Tonix’s vaccine platform has been selected by NIH’s Project NextGen for clinical testing
CHATHAM, N.J., June 25, 2024 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP) (Tonix or the Company), a fully-integrated biopharmaceutical company with marketed products and a pipeline of development candidates, today announced data presented at an oral Keynote Talk at the Vaccine Congress 2024 held June 24-25, 2024 in Prague, Czech Republic. A copy of the Company’s presentation is available under the Scientific Presentations tab of the Tonix website at www.tonixpharma.com.
The presentation titled, “A New Live Virus, One Dose Vaccine Platform for Mpox, Smallpox and COVID-19”, detailed the Company’s vaccine platform, including TNX-801 (horsepox, live virus) vaccine for preventing mpox (formerly known as monkeypox) and smallpox and TNX-1800 (horsepox expressing SARS-CoV-2 spike protein) for protecting against COVID-19. TNX-801 and TNX-1800 are live replicating attenuated vaccines based on horsepox that are believed to provide immune protection with better tolerability than modern vaccinia viruses. In the presentation, Tonix highlighted positive preclinical efficacy data, demonstrating that TNX-801 protected non-human primates against lethal challenge with intratracheal Clade 1 monkeypox virus3. After a single dose vaccination, TNX-801 prevented clinical disease and lesions and also decreased shedding in the mouth and lungs of non-human primates. These findings are consistent with mucosal immunity and suggest the ability to block forward transmission, similar to Dr. Edward Jenner’s vaccinia vaccine, which eradicated smallpox and kept mpox out of the human population.
“We are excited for the prospects of our live virus vaccine platform and look forward to advancing development for our vaccine candidates to prevent mpox, smallpox, COVID and other infectious diseases,” said Seth Lederman, M.D., Chief Executive Officer of Tonix. “TNX-801 combines immune protection with improved tolerability compared to other vaccines based on orthopoxviruses. TNX-801 is administered with a single dose which has advantages over two-dose regimens. We believe TNX-801 can be rapidly scaled up for manufacturing and can be distributed and stored without a costly and cumbersome ultra-cold supply chain. TNX-801 also has the potential to be used as a viral vector platform, for which recombinant versions can be developed to protect against other infectious diseases. Tonix developed TNX-1800 as a vaccine to protect against COVID-19. We are excited that TNX-1800 was selected for the U.S. National Institute of Health’s (NIH’s) Project NextGen and we look forward to providing updates on the program.”
The presentation noted that the global mpox outbreak, which commenced in 2022, has affected over 90,000 persons in countries where mpox had previously not been endemic, including Europe and the US. The spread of Clade IIb strain mpox in 2022 underscores the pandemic potential of mpox. Unlike Clade IIb mpox, the Clade I strain of mpox remains restricted to several Central African countries, including the Democratic Republic of the Congo, where it is currently endemic. Clade I mpox is typically associated with higher case fatality rates than Clade IIb mpox. According to the U.S. Centers for Disease Control and Prevention (CDC), and other experts, there is a significant risk that the deadlier Clade I strain may appear in the U.S.1,4.
Further, the presentation noted that the Bipartisan Commission on Biodefense recently highlighted the renewed dual threats of both a more virulent mpox epidemic as well as smallpox re-introduction from lab accidents or bad actors, while the National Academies of Science, in its review of smallpox preparedness, highlighted the need for new single dose vaccines against smallpox5,6.
The keynote presentation also included preclinical data for TNX-1800, demonstrating immunity and tolerability7,8. TNX-1800 was selected by the NIH’s Project NextGen for inclusion in clinical trials as part of a select group of next generation COVID-19 vaccine candidates with the intent to identify promising vaccine platforms. NIH plans to conduct a Phase 1 trial and cover the full cost, while Tonix provides the vaccine candidate.
Tonix Pharmaceuticals Holding Corp.*
Tonix is a fully-integrated biopharmaceutical company focused on developing, licensing and commercializing therapeutics to treat and prevent human disease and alleviate suffering. Tonix’s development portfolio is focused on central nervous system (CNS) disorders. Tonix’s priority is to submit a New Drug Application (NDA) to the FDA in the second half of 2024 for Tonmya**, a product candidate for which two statistically significant Phase 3 studies have been completed for the management of fibromyalgia. TNX-102 SL is also being developed to treat acute stress reaction as well as fibromyalgia-type Long COVID. Tonix’s CNS portfolio includes TNX-1300 (cocaine esterase), a biologic designed to treat cocaine intoxication that has Breakthrough Therapy designation. Tonix’s immunology development portfolio consists of biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases. Tonix also has product candidates in development in the areas of rare disease and infectious disease. Tonix Medicines, our commercial subsidiary, markets Zembrace® SymTouch® (sumatriptan injection) 3 mg and Tosymra® (sumatriptan nasal spray) 10 mg for the treatment of acute migraine with or without aura in adults.
*Tonix’s product development candidates are investigational new drugs or biologics and have not been approved for any indication.
**Tonmya™ is conditionally accepted by the U.S. Food and Drug Administration (FDA) as the tradename for TNX-102 SL for the management of fibromyalgia. Tonmya has not been approved for any indication.
1McQuiston JH, et al. U.S. Preparedness and Response to Increasing Clade I Mpox Cases in the Democratic Republic of the Congo. 2024, MMWR Morb Mortal Wkly Rep: United States. p. 435-440
2CDC. 2022-2023 Mpox: US Map and Case Count. https://www.cdc.gov/poxvirus/mpox/response/2022/us-map.html
3Noyce RS, et al. Viruses. 2023 Jan 26;15(2):356. doi: 10.3390/v15020356. PMID: 36851570; PMCID: PMC9965234
4World Health OrganizationSAGE meeting highlights on updated mpox vaccine recommendations. 2024, March
5Bipartisan Commission on Biofence. Box the Pox: Redicing the risk of Smallpox and Other Orthopoxviruses, Washington:2024
6U.S. National Academies of Science. Future State of Smallpox Medical Countermeasures. Washington:2024
7Awasthi M, et al. Viruses. 2023 Oct 21;15(10):2131. doi: 10.3390/v15102131. PMID: 37896908; PMCID: PMC10612059.
8Awashti AM et al Vaccines (Basel). 2023 Nov 2;11(11):1682. doi:10.3390/vaccines11111682.PMID: 38006014
Zembrace SymTouch and Tosymra are registered trademarks of Tonix Medicines. All other marks are property of their respective owners.
This press release and further information about Tonix can be found at www.tonixpharma.com.
Forward Looking Statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; risks related to the failure to successfully market any of our products; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2023, as filed with the Securities and Exchange Commission (the “SEC”) on April 1, 2024, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
Investor Contact
Jessica Morris
Tonix Pharmaceuticals
[email protected]
(862) 904-8182
Peter Vozzo
ICR Westwicke
[email protected]
(443) 213-0505
Media Contact
Katie Dodge
LaVoieHealthScience
[email protected]
(978) 360-3151

Source: Tonix Pharmaceuticals Holding Corp.
Released June 25, 2024