Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Side Effect Rates Were Low. Unicycive announced top-line results from its pivotal study to determine OLC (oxylanthanum carbonate) tolerability, safety, and dosing. The trial met its tolerability and safety endpoints with data that compares favorably with Fosrenol (lanthanum carbonate). Over 90% of the patients were able to lower their serum phosphate to target levels, with 70% reaching target at the lowest dose tested. An NDA filing is expected in 3Q24.
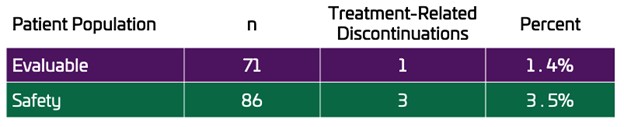
Low Discontinuation Rate Met The Primary Endpoint. Tolerability, the rate of discontinuations due to treatment related adverse events (TRAEs), was the primary endpoint. The Evaluable Population of 71 patients had only 1 TRAE discontinuation, a rate of 1.4%. In the Safety Population, a total of 3 patients out of 86 discontinued due to TRAEs, a rate of 3.5%. In total, 5 patients discontinued due to AEs in the Safety Population, 3 were related to OLC and 2 were deemed unrelated to OLC.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Tonix’s live virus vaccine TNX-801 is designed to provide long-term protection from mpox and smallpox with one dose
TNX-801 vaccination demonstrated efficacy in protecting animals from lethal challenge with intratracheal monkeypox
Clade II mpox is now endemic in the U.S. with >30,000 cases reported since May 20221 and Clade I mpox is endemic in the Democratic Republic of the Congo2
Tonix’s vaccine platform has been selected by NIH’s Project NextGen for clinical testing
CHATHAM, N.J., June 25, 2024 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP) (Tonix or the Company), a fully-integrated biopharmaceutical company with marketed products and a pipeline of development candidates, today announced data presented at an oral Keynote Talk at the Vaccine Congress 2024 held June 24-25, 2024 in Prague, Czech Republic. A copy of the Company’s presentation is available under the Scientific Presentations tab of the Tonix website at www.tonixpharma.com.
The presentation titled, “A New Live Virus, One Dose Vaccine Platform for Mpox, Smallpox and COVID-19”, detailed the Company’s vaccine platform, including TNX-801 (horsepox, live virus) vaccine for preventing mpox (formerly known as monkeypox) and smallpox and TNX-1800 (horsepox expressing SARS-CoV-2 spike protein) for protecting against COVID-19. TNX-801 and TNX-1800 are live replicating attenuated vaccines based on horsepox that are believed to provide immune protection with better tolerability than modern vaccinia viruses. In the presentation, Tonix highlighted positive preclinical efficacy data, demonstrating that TNX-801 protected non-human primates against lethal challenge with intratracheal Clade 1 monkeypox virus3. After a single dose vaccination, TNX-801 prevented clinical disease and lesions and also decreased shedding in the mouth and lungs of non-human primates. These findings are consistent with mucosal immunity and suggest the ability to block forward transmission, similar to Dr. Edward Jenner’s vaccinia vaccine, which eradicated smallpox and kept mpox out of the human population.
“We are excited for the prospects of our live virus vaccine platform and look forward to advancing development for our vaccine candidates to prevent mpox, smallpox, COVID and other infectious diseases,” said Seth Lederman, M.D., Chief Executive Officer of Tonix. “TNX-801 combines immune protection with improved tolerability compared to other vaccines based on orthopoxviruses. TNX-801 is administered with a single dose which has advantages over two-dose regimens. We believe TNX-801 can be rapidly scaled up for manufacturing and can be distributed and stored without a costly and cumbersome ultra-cold supply chain. TNX-801 also has the potential to be used as a viral vector platform, for which recombinant versions can be developed to protect against other infectious diseases. Tonix developed TNX-1800 as a vaccine to protect against COVID-19. We are excited that TNX-1800 was selected for the U.S. National Institute of Health’s (NIH’s) Project NextGen and we look forward to providing updates on the program.”
The presentation noted that the global mpox outbreak, which commenced in 2022, has affected over 90,000 persons in countries where mpox had previously not been endemic, including Europe and the US. The spread of Clade IIb strain mpox in 2022 underscores the pandemic potential of mpox. Unlike Clade IIb mpox, the Clade I strain of mpox remains restricted to several Central African countries, including the Democratic Republic of the Congo, where it is currently endemic. Clade I mpox is typically associated with higher case fatality rates than Clade IIb mpox. According to the U.S. Centers for Disease Control and Prevention (CDC), and other experts, there is a significant risk that the deadlier Clade I strain may appear in the U.S.1,4.
Further, the presentation noted that the Bipartisan Commission on Biodefense recently highlighted the renewed dual threats of both a more virulent mpox epidemic as well as smallpox re-introduction from lab accidents or bad actors, while the National Academies of Science, in its review of smallpox preparedness, highlighted the need for new single dose vaccines against smallpox5,6.
The keynote presentation also included preclinical data for TNX-1800, demonstrating immunity and tolerability7,8. TNX-1800 was selected by the NIH’s Project NextGen for inclusion in clinical trials as part of a select group of next generation COVID-19 vaccine candidates with the intent to identify promising vaccine platforms. NIH plans to conduct a Phase 1 trial and cover the full cost, while Tonix provides the vaccine candidate.
Tonix Pharmaceuticals Holding Corp.*
Tonix is a fully-integrated biopharmaceutical company focused on developing, licensing and commercializing therapeutics to treat and prevent human disease and alleviate suffering. Tonix’s development portfolio is focused on central nervous system (CNS) disorders. Tonix’s priority is to submit a New Drug Application (NDA) to the FDA in the second half of 2024 for Tonmya**, a product candidate for which two statistically significant Phase 3 studies have been completed for the management of fibromyalgia. TNX-102 SL is also being developed to treat acute stress reaction as well as fibromyalgia-type Long COVID. Tonix’s CNS portfolio includes TNX-1300 (cocaine esterase), a biologic designed to treat cocaine intoxication that has Breakthrough Therapy designation. Tonix’s immunology development portfolio consists of biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases. Tonix also has product candidates in development in the areas of rare disease and infectious disease. Tonix Medicines, our commercial subsidiary, markets Zembrace® SymTouch® (sumatriptan injection) 3 mg and Tosymra® (sumatriptan nasal spray) 10 mg for the treatment of acute migraine with or without aura in adults.
*Tonix’s product development candidates are investigational new drugs or biologics and have not been approved for any indication.
**Tonmya™ is conditionally accepted by the U.S. Food and Drug Administration (FDA) as the tradename for TNX-102 SL for the management of fibromyalgia. Tonmya has not been approved for any indication.
1McQuiston JH, et al. U.S. Preparedness and Response to Increasing Clade I Mpox Cases in the Democratic Republic of the Congo. 2024, MMWR Morb Mortal Wkly Rep: United States. p. 435-440 2CDC. 2022-2023 Mpox: US Map and Case Count. https://www.cdc.gov/poxvirus/mpox/response/2022/us-map.html 3Noyce RS, et al. Viruses. 2023 Jan 26;15(2):356. doi: 10.3390/v15020356. PMID: 36851570; PMCID: PMC9965234 4World Health OrganizationSAGE meeting highlights on updated mpox vaccine recommendations. 2024, March 5Bipartisan Commission on Biofence. Box the Pox: Redicing the risk of Smallpox and Other Orthopoxviruses, Washington:2024 6U.S. National Academies of Science. Future State of Smallpox Medical Countermeasures. Washington:2024 7Awasthi M, et al. Viruses. 2023 Oct 21;15(10):2131. doi: 10.3390/v15102131. PMID: 37896908; PMCID: PMC10612059. 8Awashti AM et al Vaccines (Basel). 2023 Nov 2;11(11):1682. doi:10.3390/vaccines11111682.PMID: 38006014
Zembrace SymTouch and Tosymra are registered trademarks of Tonix Medicines. All other marks are property of their respective owners.
This press release and further information about Tonix can be found at www.tonixpharma.com.
Forward Looking Statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; risks related to the failure to successfully market any of our products; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2023, as filed with the Securities and Exchange Commission (the “SEC”) on April 1, 2024, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
– Successfully Established Favorable Tolerability and Safety of OLC –
– New Drug Application (NDA) Submission Anticipated in Q3 2024 –
– Webcast and Conference Call Today at 8:30 A.M. ET –
LOS ALTOS, Calif., June 25, 2024 (GLOBE NEWSWIRE) — Unicycive Therapeutics, Inc. (Nasdaq: UNCY), a clinical-stage biotechnology company developing therapies for patients with kidney disease (the “Company” or “Unicycive”), today announced positive results from the Oxylanthanum Carbonate (OLC) UNI-OLC-201 pivotal clinical trial with regard to both safety and tolerability endpoints. OLC is a next-generation lanthanum-based phosphate binding agent utilizing proprietary nanoparticle technology being developed for the treatment of hyperphosphatemia in patients with chronic kidney disease (CKD).
The study established promising tolerability of OLC at clinically effective doses in CKD patients on hemodialysis. In terms of tolerability, OLC had a low rate of discontinuation due to adverse events (AEs) with only 5/86 patients (6%) discontinuing from the Study. Of the 5 discontinuations, 3 were treatment-related and 2 were not related to treatment. Importantly, the Company believes the low discontinuation rate for OLC compares favorably to a discontinuation rate due to AEs of 14% for Fosrenol® from its U.S. Food and Drug Administration (FDA)-approved Package Insert.
The primary endpoint was defined as the rate of discontinuations due to treatment-related AEs leading to discontinuation in the maintenance period. In the UNI-OLC-201 trial, there was only 1 discontinuation due to a treatment-related AE in the Evaluable Population (n=71), a rate of 1.4%. In the full Safety Population, a total of 3 patients discontinued due to treatment-related AEs, a rate of 3.5%.
The secondary endpoint assessing safety was also favorable as most treatment-related AEs were mild to moderate in severity and there were no treatment-related serious adverse events (SAEs) reported in the Safety Population. The treatment-related AEs reported in ≥5% of patients were diarrhea (9%) and vomiting (6%) which also compares favorably to Fosrenol and other phosphate binders on the market.
While the study was not designed to evaluate efficacy, the trial enrolled patients on stable doses of approved hyperphosphatemia medications. At baseline approximately 59% of patients had phosphate levels ≤5.5 mg/dL, the level recommended by KDOQI guidelines. After washout from the prior phosphate binders, 90% of patients were able to achieve phosphate levels ≤5.5ng/dL at the end of titration with OLC.
“We are immensely pleased with the outcome of the UNI-OLC-201 clinical trial as the results demonstrate extremely promising tolerability results in real-world dialysis patients,” said Shalabh Gupta, MD, Chief Executive Officer of Unicycive. “The study was well received by investigators and patients as we were able to successfully over-enroll the study with 71 evaluable patients. In addition, we were able to obtain phosphate control in 90% of the Safety Population during titration and OLC proved to be highly potent at lower doses. We would like to thank our investigators, study coordinators, and the patients who dedicated their time and energy for our clinical trial.”
Dr. Gupta continued, “We believe these favorable results confirming tolerability for OLC are the final data component needed to support submission of a New Drug Application to the FDA utilizing the 505(b)(2) regulatory pathway. The submission package will also include the previously disclosed preclinical data and the data establishing bioequivalence to Fosrenol. The encouraging performance of OLC gives us a high degree of confidence and provides potential clinical validation of OLC’s best-in-class commercial promise for patients suffering from hyperphosphatemia.”
“Based on real world evidence, approximately 40% of patients on dialysis are unable to achieve adequate serum phosphate control as defined by established KDOQI treatment guidelines1. The UNI-OLC-201 study was representative of the U.S. dialysis patient population. Uncontrolled hyperphosphatemia is an important problem for patients and physicians because it can lead to other major complications including cardiovascular disease. I am encouraged by the results from the OLC pivotal trial, and I believe that a product like OLC that improves phosphate control and reduces the number of pills could have a meaningful impact on the overall care of CKD patients on dialysis,” added Pablo Pergola, MD, PhD, Research Director, Clinical Advancement Center, Renal Associates, P.A., and principal investigator for the UNI-OLC-201 trial.
Conference Call & Webcast Details
Unicycive will host a webcast and conference call with accompanying slides today at 8:30 a.m. ET. The live and archived webcast may be accessed on the Unicycive website under the Investors section: Events and Presentations. The live call can be accessed by dialing +1 (646) 876-9923 with Meeting ID: 96518079674 and Passcode: 273069.
Presentation slides will be provided at the start of the conference call on the Unicycive website under the Investors section: Events and Presentations.
OLC-201 Data Summary
Overview
The results from the trial are focused on two patient populations: the full Safety Population and the Evaluable Population. The Safety Population (n=86) included all patients who entered titration and received at least one dose of OLC. The Evaluable Population (n=71) required a patient to have a serum phosphate level of ≤5.5 mg/dL at the end of titration and received at least one dose of OLC in the maintenance period.
Once patients were enrolled into the trial, they went through a washout period for two weeks to clear their current phosphate binder from the body. Participants were initially dosed at 500 mg of OLC three times a day (TID) and titrated to a clinically effective dose that is defined as the dose required to achieve a serum phosphate level of ≤5.5 mg/dL. The maximum dose of OLC tested was 3000 mg/day (1000 mg TID). As a reminder, all approved phosphate binders are administered on a dose titration schedule based on the control of serum phosphate. Once titrated to a clinically effective dose, patients were then treated in a maintenance period with OLC for four weeks to evaluate tolerability.
Demographics and Enrollment Summary
In the study, 106 patients were enrolled, of which 86 patients entered titration and were followed as the Safety Population. Of the 86, 78 entered the maintenance period. Of the 78 patients that entered maintenance, 7 patients did not have phosphate control, leaving an Evaluable Population of 71 patients, exceeding the planned enrollment number of 60. Of the 86 patients, the trial enrolled 47 males and 39 females with a mean age of 62. Renvela® was the most prescribed phosphate binder for patients entering the study.
Primary Endpoint – Tolerability:
The objective of the OLC-201 trial was to evaluate the tolerability of clinically effective doses of OLC in CKD patients on dialysis. A clinically effective dose was established when a patient achieved a serum phosphate level ≤5.5 mg/dL. Tolerability was assessed based on the incidence of treatment-related AEs leading to discontinuation from the study in the maintenance period. In the OLC-201 trial, there was only 1 discontinuation due to a treatment-related AE in the Evaluable Population, a rate of 1.4%. In the Safety Population of 86 patients there were only 3 treatment-related discontinuations, a rate of 3.5%. In total, 5 patients discontinued due to AEs in the Safety Population, 3 were related to OLC and 2 were deemed unrelated to OLC.
Secondary Endpoint – Safety:
The secondary endpoint assessing safety was reported as the treatment-related AEs occurring in ≥5% of patients. The safety analysis covered all 86 patients in the Safety Population. Consistent with the AEs observed with other phosphate binders, the AEs were gastrointestinal related with diarrhea and vomiting being the most common at 9% and 6% respectively. There were no treatment-related serious adverse events (SAEs). Six patients experienced SAEs but those were deemed not related to OLC treatment. Most treatment-related AEs were mild to moderate in severity with only 2 AEs reported as severe.
Unicycive continues to assess the pharmacokinetics from this trial and those final data will be included in the NDA package.
Serum Phosphate Control
While the UNI-OLC-201 study was not designed to evaluate efficacy, the trial enrolled patients on stable doses of approved hyperphosphatemia medications. At baseline 59% of patients had phosphate levels ≤5.5 mg/dL, the level recommended by KDOQI guidelines. After washout from the prior phosphate binders, 90% of patients were able to achieve phosphate levels ≤5.5ng/dL at the end of titration with OLC. This includes the last serum phosphate levels from all patients including those that discontinued during titration: 77/86 (90%).
In addition, 69% of the 71 Evaluable Patients achieved a target serum phosphate level of ≤5.5 mg/dL at OLC doses of 1500 mg/day or lower.
About Oxylanthanum Carbonate (OLC)
Oxylanthanum carbonate is a next-generation lanthanum-based phosphate binding agent utilizing proprietary nanoparticle technology being developed for the treatment of hyperphosphatemia in patients with chronic kidney disease (CKD). OLC has over forty issued and granted patents globally. Its potential best-in-class profile may have meaningful patient adherence benefits over currently available treatment options as it requires a lower pill burden for patients in terms of number and size of pills per dose that are swallowed instead of chewed. Based on a survey conducted in 2022, Nephrologists stated that the greatest unmet need in the treatment of hyperphosphatemia with phosphate binders is a lower pill burden and better patient compliance.2 The global market opportunity for treating hyperphosphatemia is projected to be in excess of $2.5 billion in 2023, with the United States accounting for more than $1 billion of that total.3 Despite the availability of several FDA-cleared medications, 75 percent of U.S. dialysis patients fail to achieve the target phosphorus levels recommended by published medical guidelines.
Unicycive is seeking FDA approval of OLC via the 505(b)(2) regulatory pathway. As part of the clinical development program, two prior clinical studies were conducted in over 100 healthy volunteers. The first study was a dose-ranging Phase I study to determine safety and tolerability. The second study was a randomized, open-label, two-way crossover bioequivalence study to establish pharmacodynamic bioequivalence between OLC and Fosrenol. Based on the topline results of the bioequivalence study, pharmacodynamic (PD) bioequivalence of OLC to Fosrenol was established.
About Hyperphosphatemia
Hyperphosphatemia is a serious medical condition that occurs in nearly all patients with End Stage Renal Disease (ESRD). If left untreated, hyperphosphatemia leads to secondary hyperparathyroidism (SHPT), which then results in renal osteodystrophy (a condition similar to osteoporosis and associated with significant bone disease, fractures and bone pain); cardiovascular disease with associated hardening of arteries and atherosclerosis (due to deposition of excess calcium-phosphorus complexes in soft tissue). Importantly, hyperphosphatemia is independently associated with increased mortality for patients with chronic kidney disease on dialysis. Based on available clinical data to date, over 80% of patients show signs of cardiovascular calcification by the time they become dependent on dialysis.
Dialysis patients are already at an increased risk for cardiovascular disease (because of underlying diseases such as diabetes and hypertension), and hyperphosphatemia further exacerbates this. Treatment of hyperphosphatemia is aimed at lowering serum phosphate levels via two means: (1) restricting dietary phosphorus intake; and (2) using, on a daily basis, and with each meal, oral phosphate binding drugs that facilitate fecal elimination of dietary phosphate rather than its absorption from the gastrointestinal tract into the bloodstream.
About Unicycive Therapeutics
Unicycive Therapeutics is a biotechnology company developing novel treatments for kidney diseases. Unicycive’s lead drug candidate, oxylanthanum carbonate (OLC), is a novel investigational phosphate binding agent being developed for the treatment of hyperphosphatemia in chronic kidney disease patients on dialysis. UNI-494 is a patent-protected new chemical entity in clinical development for the treatment of conditions related to acute kidney injury. For more information, please visit Unicycive.com and follow us on LinkedIn, X, and YouTube.
Forward-looking statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified using words such as “anticipate,” “believe,” “forecast,” “estimated” and “intend” or other similar terms or expressions that concern Unicycive’s expectations, strategy, plans or intentions. These forward-looking statements are based on Unicycive’s current expectations and actual results could differ materially. There are several factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results; our clinical trials may be suspended or discontinued due to unexpected side effects or other safety risks that could preclude approval of our product candidates; risks related to business interruptions, which could seriously harm our financial condition and increase our costs and expenses; dependence on key personnel; substantial competition; uncertainties of patent protection and litigation; dependence upon third parties; and risks related to failure to obtain FDA clearances or approvals and noncompliance with FDA regulations. Topline data from the Oxylanthanum carbonate (OLC) pivotal trial is preliminary and subject to change based on further detailed analysis. Actual results may differ materially from those indicated by such forward-looking statements as a result of various important factors, including: the uncertainties related to market conditions and other factors described more fully in the section entitled ‘Risk Factors’ in Unicycive’s Annual Report on Form 10-K for the year ended December 31, 2023, and other periodic reports filed with the Securities and Exchange Commission. Any forward-looking statements contained in this press release speak only as of the date hereof, and Unicycive specifically disclaims any obligation to update any forward-looking statement, whether as a result of new information, future events or otherwise.
Fosrenol® (lanthanum carbonate) is a registered trademark of Shire International Licensing BV. Renvela® (sevelamer carbonate) is a registered trademark of Sanofi.
1KDOQI treatment guidelines 2Reason Research, LLC 2022 company sponsored survey. Results here. 3Fortune Business InsightsTM, Hyperphosphatemia Treatment Market, 2021-2028
In a bold move that underscores the burgeoning demand for weight loss and diabetes treatments, Danish pharmaceutical giant Novo Nordisk has announced a monumental $4.1 billion investment to construct a state-of-the-art manufacturing facility in Clayton, North Carolina. This strategic decision marks a significant escalation in the company’s commitment to increasing the supply of its blockbuster drugs, Wegovy and Ozempic, which have taken the medical world by storm.
The new 1.4 million-square-foot plant, slated for completion between 2027 and 2029, will be dedicated to the crucial tasks of filling and packaging syringes and injection pens for these game-changing medications. This expansion is not just about bricks and mortar; it represents a transformative step in Novo Nordisk’s production capabilities and its position in the competitive pharmaceutical landscape.
The timing of this investment is critical. Wegovy and Ozempic, both part of a class of drugs known as GLP-1s, have seen demand skyrocket, outpacing the company’s current production capacity. The resulting shortages have left many patients struggling to access these treatments, which have shown remarkable efficacy in managing weight and diabetes. The new facility aims to bridge this gap, potentially revolutionizing access to these sought-after therapies.
The scale of Novo Nordisk’s commitment is evident in the numbers. The company plans to invest a staggering $6.8 billion in production this year alone, a significant increase from the $4 billion invested last year. This ramping up of investment reflects not just the current demand but also the company’s bullish outlook on the future of these treatments.
The impact of this expansion extends beyond the realm of healthcare. The new facility is set to create 1,000 new jobs, adding to the 2,500 employees already working at Novo Nordisk’s existing North Carolina plants. This influx of high-quality jobs represents a significant economic boon for the region, further cementing North Carolina’s status as a hub for pharmaceutical manufacturing.
The demand for Wegovy, in particular, underscores the potential market for effective weight loss treatments. With an average of 35,000 U.S. patients starting Wegovy each week – up from 27,000 in May – the drug has clearly struck a chord in a nation grappling with an obesity epidemic. However, the current shortage of lower doses has hampered the drug’s rollout, a problem this new facility aims to address.
Novo Nordisk’s expansion is not happening in isolation. The weight loss and diabetes treatment market has become a battleground for pharmaceutical giants, with companies like Eli Lilly also investing heavily in manufacturing capacity for similar drugs. This competition is likely to drive further innovation and potentially lead to more accessible treatments for patients in the future.
The Clayton facility will join Novo Nordisk’s existing manufacturing network, which includes plants in Denmark, France, China, Japan, and several other countries. However, this significant investment in U.S. manufacturing capacity signals the company’s recognition of the importance of the American market and its commitment to serving patients in the region.
As construction begins on this new facility, the pharmaceutical industry watches with keen interest. The success of this venture could set a new standard for production capacity in the industry and potentially reshape how companies approach the manufacturing of high-demand drugs.
In the grand scheme of things, Novo Nordisk’s $4.1 billion investment is more than just an expansion of manufacturing capacity. It represents a vote of confidence in the future of weight loss and diabetes treatments, a commitment to addressing critical healthcare needs, and a strategic move to solidify the company’s position as a leader in this rapidly evolving field. As the facility takes shape over the coming years, it may well become a symbol of the transformative power of targeted pharmaceutical investment in addressing global health challenges.
In a strategic move to bolster its position in the rare disease and ophthalmology markets, ANI Pharmaceuticals has announced its acquisition of Alimera Sciences for approximately $381 million. This transformative deal, expected to close in the third quarter of 2024, marks a significant step in ANI’s growth strategy and expansion into the global pharmaceutical landscape.
The acquisition terms include an upfront payment of $5.50 per share in cash, representing a substantial 75% premium over Alimera’s recent closing price. Additionally, Alimera shareholders will receive a contingent value right (CVR) of up to $0.50 per share, tied to the achievement of specific revenue targets in 2026 and 2027. This structure aligns the interests of both companies and incentivizes future growth.
At the heart of this acquisition are Alimera’s two key commercial products, ILUVIEN® and YUTIQ®, both targeting eye conditions such as diabetic macular edema and chronic non-infectious uveitis. These assets are expected to contribute significantly to ANI’s revenue stream, adding approximately $105 million in highly durable branded revenue. The integration of these products aligns with ANI’s recent strategic focus on ophthalmology, complementing its existing rare disease portfolio.
The deal is projected to have a substantial positive impact on ANI’s financial performance. The company anticipates high single-digit to low double-digit accretion in adjusted non-GAAP earnings per share (EPS) in 2025, with even more substantial accretion expected in subsequent years. Furthermore, ANI projects an additional $35-$38 million in adjusted non-GAAP EBITDA for 2025, including approximately $10 million in identified cost synergies.
Beyond the immediate financial benefits, this acquisition significantly expands ANI’s geographic footprint. Alimera’s established presence in European markets, including direct operations in Germany, the United Kingdom, Portugal, and Ireland, provides ANI with a springboard for international growth. The deal also brings valuable partnerships in Asia and the Middle East, further diversifying ANI’s global reach.
Strategically, this move strengthens ANI’s position in the rare disease sector, which is expected to become the company’s primary growth driver. Post-acquisition, the rare disease segment is projected to account for approximately 45% of ANI’s pro forma 2024 revenues, with robust growth potential. The transaction also leverages ANI’s existing rare disease infrastructure, creating operational efficiencies and expanding its reach to over 3,600 physicians in the ophthalmology field.
To finance the acquisition, ANI will utilize a combination of cash on hand and $280 million in committed debt financing from J.P. Morgan and Blackstone Credit & Insurance. The company anticipates a pro-forma leverage of 3.2x upon closing, with significant organic de-levering expected in 2025.
The boards of directors of both companies have approved the transaction, which now awaits customary closing conditions, including regulatory approvals and Alimera shareholder approval. Both companies have enlisted top-tier financial and legal advisors to navigate the complexities of the deal, underscoring its strategic importance.
This acquisition represents a pivotal moment for ANI Pharmaceuticals, positioning it as a stronger player in the rare disease and ophthalmology markets. By integrating Alimera’s products and expertise, ANI is set to enhance its market presence, diversify its revenue streams, and potentially accelerate the growth of its existing products, including Purified Cortrophin® Gel, in the ophthalmology sector.
As the pharmaceutical industry continues to evolve, with an increasing focus on specialized treatments for rare diseases, this strategic move by ANI Pharmaceuticals demonstrates its commitment to growth and innovation. The successful integration of Alimera Sciences could serve as a catalyst for ANI’s long-term success, benefiting patients, physicians, and shareholders alike in the rapidly advancing field of ophthalmology and rare disease treatment.
• Established Medium Dose as Safe and Tolerable Dose in Current OCU410ST Clinical Trial • DSMB Determination to Proceed with High Dose Cohort Dosing
MALVERN, Pa., June 21, 2024 (GLOBE NEWSWIRE) — Ocugen, Inc. (Ocugen or the Company) (NASDAQ: OCGN), a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines, today announced that the Data and Safety Monitoring Board (DSMB) for the OCU410ST GARDian clinical trial recently convened and approved to proceed with dosing the high dose of OCU410ST in the dose-escalation phase of the study. OCU410ST (AAV5-hRORA) is a modifier gene therapy candidate being developed for Stargardt disease. Stargardt disease affects approximately 100,000 people in the U.S. and Europe combined.
Six patients with Stargardt disease have been dosed in the Phase 1/2 clinical trial to date in the low dose cohort and medium dose cohort. An additional three patients will be dosed with the high dose in cohort 3.
“The DSMB has recommended moving forward to dose subsequent subjects with Stargardt disease at the targeted high dose,” said Dr. Peter Y. Chang, MD, FACS, DSMB Chair for the OCU410ST clinical trial. “No serious adverse events (SAEs) related to OCU410ST have been reported to date. This is an important next step in the clinical progress for OCU410ST and encouraging for patients living with this most common form of inherited retinal disease.”
“We are delighted to report a second positive DSMB recommendation for the treatment of Stargardt disease and build upon the favorable safety and tolerability profile exhibited by OCU410ST,” said Huma Qamar, M.D., MPH, Chief Medical Officer of Ocugen. “We recognize the high unmet medical need for Stargardt patients as there is no approved product. We are enthusiastic about OCU410ST as a potential one-time treatment for life with a single sub-retinal injection. We look forward to sharing a clinical trial update later this year.”
The Phase 1/2 GARDian clinical trial will include up to 42 subjects—30 adults and 12 children with Stargardt disease—who exhibit mild to moderate disease symptoms and will assess the safety of unilateral subretinal administration of OCU410ST. The clinical trial is being conducted in two phases. Phase 1 is a multicenter, open-label, dose-ranging/dose-escalation study consisting of three dose levels [low dose (3.75× 1010 vg/mL), medium dose (7.5× 1010 vg/mL), and high dose (2.25× 1011 vg/mL)]. Phase 2 is a randomized, outcome accessor-blinded, dose-expansion study in which adult and pediatric subjects will be enrolled in a 1:1:1 ratio to randomize subjects into two different treatment groups at varying dose levels, or a control (untreated group), allowing for a comprehensive assessment of the treatment’s efficacy across different dosages.
Currently, patients with Stargardt disease have no FDA-approved therapeutic options. Ocugen is dedicated to providing a gene-agnostic treatment approach for patients living with inherited retinal diseases and is encouraged that the Phase 1/2 GARDian trial for OCU410ST remains on track.
About Stargardt Disease
Stargardt disease is a genetic eye disorder that causes retinal degeneration and vision loss. Stargardt disease is the most common form of inherited macular degeneration. The progressive vision loss associated with Stargardt disease is caused by the degeneration of photoreceptor cells in the central portion of the retina called the macula.
Decreased central vision due to loss of photoreceptors in the macula is the hallmark of Stargardt disease. Some peripheral vision is usually preserved. Stargardt disease typically develops during childhood or adolescence, but the age of onset and rate of progression can vary. The retinal pigment epithelium (RPE), a layer of cells supporting photoreceptors, is also affected in people with Stargardt disease.
About OCU410ST
OCU410ST utilizes an AAV delivery platform for the retinal delivery of the RORA (RAR Related Orphan Receptor A) gene. It represents Ocugen’s modifier gene therapy approach, which is based on Nuclear Hormone Receptor (NHR) RORA that regulates pathway links to Stargardt disease such as lipofuscin formation, oxidative stress, complement formation, inflammation, and cell survival networks.
About Ocugen, Inc. Ocugen, Inc. is a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines that improve health and offer hope for patients across the globe. We are making an impact on patient’s lives through courageous innovation—forging new scientific paths that harness our unique intellectual and human capital. Our breakthrough modifier gene therapy platform has the potential to treat multiple retinal diseases with a single product, and we are advancing research in infectious diseases to support public health and orthopedic diseases to address unmet medical needs. Discover more at www.ocugen.com and follow us on X and LinkedIn.
Cautionary Note on Forward-Looking Statements This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995, including, but not limited to, statements regarding qualitative assessments of available data, potential benefits, expectations for ongoing clinical trials, anticipated regulatory filings and anticipated development timelines, which are subject to risks and uncertainties. We may, in some cases, use terms such as “predicts,” “believes,” “potential,” “proposed,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Such statements are subject to numerous important factors, risks, and uncertainties that may cause actual events or results to differ materially from our current expectations, including, but not limited to, the risks that preliminary, interim and top-line clinical trial results may not be indicative of, and may differ from, final clinical data; that unfavorable new clinical trial data may emerge in ongoing clinical trials or through further analyses of existing clinical trial data; that earlier non-clinical and clinical data and testing of may not be predictive of the results or success of later clinical trials; and that that clinical trial data are subject to differing interpretations and assessments, including by regulatory authorities. These and other risks and uncertainties are more fully described in our periodic filings with the Securities and Exchange Commission (SEC), including the risk factors described in the section entitled “Risk Factors” in the quarterly and annual reports that we file with the SEC. Any forward-looking statements that we make in this press release speak only as of the date of this press release. Except as required by law, we assume no obligation to update forward-looking statements contained in this press release whether as a result of new information, future events, or otherwise, after the date of this press release.
Tonix is a clinical-stage biopharmaceutical company focused on discovering, licensing, acquiring and developing therapeutics and diagnostics to treat and prevent human disease and alleviate suffering. Tonix’s portfolio is composed of immunology, rare disease, infectious disease, and central nervous system (CNS) product candidates. Tonix’s immunology portfolio includes biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-15001 which is a humanized monoclonal antibody targeting CD40-ligand being developed for the prevention of allograft and xenograft rejection and for the treatment of autoimmune diseases. A Phase 1 study of TNX-1500 is expected to be initiated in the second half of 2022. Tonix’s rare disease portfolio includes TNX-29002 for the treatment of Prader-Willi syndrome. TNX-2900 has been granted Orphan-Drug Designation by the FDA. Tonix’s infectious disease pipeline includes a vaccine in development to prevent smallpox and monkeypox called TNX-8013, next-generation vaccines to prevent COVID-19, and an antiviral to treat COVID-19. Tonix’s lead vaccine candidates for COVID-19 are TNX-1840 and TNX-18504, which are live virus vaccines based on Tonix’s recombinant pox vaccine (RPV) platform. TNX-35005 (sangivamycin, i.v. solution) is a small molecule antiviral drug to treat acute COVID-19 and is in the pre-IND stage of development. TNX-102 SL6, (cyclobenzaprine HCl sublingual tablets), is a small molecule drug being developed to treat Long COVID, a chronic post-acute COVID-19 condition. Tonix expects to initiate a Phase 2 study in Long COVID in the second quarter of 2022. The Company’s CNS portfolio includes both small molecules and biologics to treat pain, neurologic, psychiatric and addiction conditions. Tonix’s lead CNS candidate, TNX-102 SL, is in mid-Phase 3 development for the management of fibromyalgia with a new Phase 3 study launched in the second quarter of 2022. Finally, TNX-13007 is a biologic designed to treat cocaine intoxication that is expected to start a Phase 2 trial in the second quarter of 2022. TNX-1300 has been granted Breakthrough Therapy Designation by the FDA.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
NDA For Tonmya Expected In 2H24. Tonix announced that it has received written feedback from the FDA from its pre-NDA meetings. The meetings covered the CMC (chemistry, manufacturing, and controls) section of the Tonmya new drug application (NDA). Based on the written feedback, Tonix believes it is in alignment with the FDA on important issues. This keeps the NDA filing on schedule for submission in 2H24.
Tonix Reaches CMC Agreement and Alignment. Topics of the meetings included the proposed drug substance and commercial specifications, shelf-life assignment, manufacturing, and commercial drug packaging. We see this as a significant milestone toward the NDA submission and approval, as many NDA applications have been delayed or received Complete Response Letters (CRLs) due to issues with manufacturing and the CMC section.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Cocrystal Pharma, Inc. is a clinical-stage biotechnology company discovering and developing novel antiviral therapeutics that target the replication process of influenza viruses, coronaviruses (including SARS-CoV-2), hepatitis C viruses and noroviruses. Cocrystal employs unique structure-based technologies and Nobel Prize-winning expertise to create first- and best-in-class antiviral drugs. For further information about Cocrystal, please visit www.cocrystalpharma.com.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
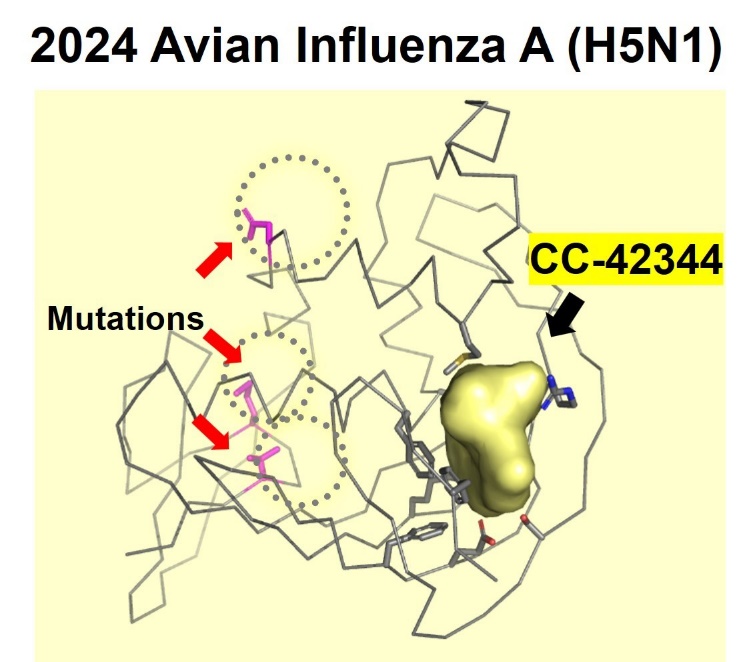
Potential Efficacy Against A New Strain Of Avian Influenza. Cocrystal has announced that studies of its influenza drug, CC-42344, have shown efficacy against the new avian influenza strain H5N1. Cocrystal has used its proprietary technology to determine the strain’s mutations and structure, then conduct preliminary tests. The binding site for CC-42344 was not changed and can block reproduction of the new strain, making it an effective vaccine against the virus.
Avian Influenza Strain Has Begun To Infect Humans. Avian influenza H5N1 has caused significant illness in commercial bird flocks since 2003. Its impact was limited until recent outbreaks in dairy cattle and infections in diary workers. This ability to mutate and infect other species is a significant step toward causing widespread outbreaks in humans, elevating it to a public health concern.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Tonix also has completed the second and final pre-New Drug Application (NDA) meeting and discussed nonclinical, clinical pharmacology and clinical matters with the FDA, formal minutes pending
On track to submit NDA to the FDA in the second half of 2024
CHATHAM, N.J., June 20, 2024 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP) (Tonix or the Company), a fully-integrated biopharmaceutical company with marketed products and a pipeline of development candidates, today announced receipt of the formal minutes from a recent pre-New Drug Application (NDA) Type-B Chemistry, Manufacturing, and Controls (CMC) meeting with the U.S. Food and Drug Administration (FDA) for Tonmya™ for the management of fibromyalgia. The purpose of the meeting was to seek alignment and agreement with the FDA on key CMC topics to support a planned NDA submission for Tonmya for the management of fibromyalgia. Based on formal written feedback, the Company believes it is aligned with the FDA on key topics, including proposed drug substance and drug product commercial specifications, shelf life assignment, manufacturing and commercial drug packaging.
In addition, the Company has completed the second and final pre-NDA meeting for Tonmya with the FDA and discussed nonclinical, clinical pharmacology and clinical matters. The Company awaits formal minutes after which it expects to announce the results of that meeting. The Company remains on track to submit the NDA for Tonmya for the management of fibromyalgia to the FDA in the second half of 2024.
“We remain encouraged and excited about the prospect of bringing the first new treatment to market for fibromyalgia patients in over a decade, and this encouraging meeting with the FDA is an important milestone as we head into the final stages of completion of the NDA package,” said Seth Lederman, M.D., Chief Executive Officer of Tonix Pharmaceuticals. “During the pre-NDA CMC meeting, the FDA affirmed alignment with Tonix on CMC content and commercial strategy for Tonmya, and we are very appreciative of the FDA’s guidance as we prepare for our NDA submission. We are currently actively preparing a dual manufacturing launch strategy with global contract development and manufacturing organization (CDMO) Almac Pharma Services and another CDMO.”
About Fibromyalgia
Fibromyalgia is a chronic pain disorder that is understood to result from amplified sensory and pain signaling within the central nervous system. Fibromyalgia afflicts an estimated 6 million to 12 million adults in the U.S., the majority of whom are women. Symptoms of fibromyalgia include chronic widespread pain, nonrestorative sleep, fatigue, and morning stiffness. Other associated symptoms include cognitive dysfunction and mood disturbances, including anxiety and depression. Individuals suffering from fibromyalgia struggle with their daily activities, have impaired quality of life, and frequently are disabled. Physicians and patients report common dissatisfaction with currently marketed products.
About Tonmya* (also known as TNX-102 SL)
Tonmya is a centrally acting, non-opioid, non-addictive, bedtime medication. The tablet is a patented sublingual formulation of cyclobenzaprine hydrochloride developed for the management of fibromyalgia. In December 2023, the company announced highly statistically significant and clinically meaningful topline results in RESILIENT, the second pivotal Phase 3 clinical trial of Tonmya for the management of fibromyalgia. In the study, Tonmya met its pre-specified primary endpoint, significantly reducing daily pain compared to placebo (p=0.00005) in participants with fibromyalgia. Statistically significant and clinically meaningful results were also seen in all six key secondary endpoints related to improving sleep quality, reducing fatigue and improving overall fibromyalgia symptoms and function. RELIEF, the first statistically significant Phase 3 trial of Tonmya in fibromyalgia, was completed in December 2020. It met its pre-specified primary endpoint of daily pain reduction compared to placebo (p=0.010) and showed activity in key secondary endpoints.
*Tonmya™ is conditionally accepted by the U.S. Food and Drug Administration as the tradename for TNX-102 SL for the management of fibromyalgia. Tonmya has not been approved for any indication.
Tonix Pharmaceuticals Holding Corp.*
Tonix is a biopharmaceutical company focused on developing, licensing and commercializing therapeutics to treat and prevent human disease and alleviate suffering. Tonix’s development portfolio is focused on central nervous system (CNS) disorders. Tonix’s priority is to submit a New Drug Application (NDA) to the FDA in the second half of 2024 for Tonmya1, a product candidate for which two statistically significant Phase 3 studies have been completed for the management of fibromyalgia. TNX-102 SL is also being developed to treat acute stress reaction as well as fibromyalgia-type Long COVID. Tonix’s CNS portfolio includes TNX-1300 (cocaine esterase), a biologic designed to treat cocaine intoxication that has FDA Breakthrough Therapy designation that is in Phase 2 development and is supported by a grant from the National Institute of Drug Abuse. Tonix’s immunology development portfolio consists of biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases. Tonix also has product candidates in development in the areas of rare disease and infectious disease. Tonix Medicines, our commercial subsidiary, markets Zembrace® SymTouch® (sumatriptan injection) 3 mg and Tosymra® (sumatriptan nasal spray) 10 mg for the treatment of acute migraine with or without aura in adults.
*Tonix’s product development candidates are investigational new drugs or biologics and have not been approved for any indication.
1Tonmya™ is conditionally accepted by the U.S. Food and Drug Administration (FDA) as the tradename for TNX-102 SL for the management of fibromyalgia. Tonmya has not been approved for any indication.
Zembrace SymTouch and Tosymra are registered trademarks of Tonix Medicines. All other marks are property of their respective owners.
This press release and further information about Tonix can be found at www.tonixpharma.com.
Forward Looking Statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; risks related to the failure to successfully market any of our products; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2023, as filed with the Securities and Exchange Commission (the “SEC”) on April 1, 2024, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
BOTHELL, Wash., June 20, 2024 (GLOBE NEWSWIRE) — Cocrystal Pharma, Inc.’s (Nasdaq: COCP) novel, broad-spectrum antiviral CC-42344 inhibits activity in the highly pathogenic avian influenza A (H5N1) PB2 protein recently identified in infected dairy cattle, according to recently completed in vitro studies. CC-42344 is a new class of antiviral drugs designed to block essential steps in the replication and transcription of the influenza A virus.
Cocrystal demonstrated the potential efficacy of CC-42344 against the new avian flu strain with the recently published genome sequence for H5N1. Using its proprietary structure-based platform, Cocrystal created a high-resolution crystal structure of this avian PB2 protein and confirmed that CC-42344 binds to its highly conserved PB2 region. The in vitro data were generated testing CC-42344 against the avian H5N1 PB2 protein and further support CC-42344’s activity similar to that of Cocrystal’s data with pandemic and seasonal influenza A.
CC-42344 binds to the highly conserved region of the avian influenza A H5N1 PB2 protein
“The findings validate our broad-spectrum approach to the treatment and prevention of pandemic flu. This is important as there are no specific FDA-approved vaccines to prevent infections by this virus in humans,” said Sam Lee, PhD, President and co-CEO of Cocrystal. “These findings support our previously reported preclinical data showing that CC-42344 is highly active against seasonal and pandemic influenza A strains, including emerging mutations. CC-42344 is an inhibitor compound providing a unique mechanism of action with a high barrier to resistance.”
“Recent CDC reported avian flu outbreaks in the U.S., which include the first cases of humans exposed to infected dairy cows, are concerning,” said James Martin, CFO and co-CEO of Cocrystal. “The CDC reported three additional cases of avian influenza infection from exposure to dairy cows in early June and avian flu is now confirmed in more than 100 dairy herds in 12 U.S. states.”
About Avian Influenza A H5N1 Avian influenza A H5N1 was reported in 889 cases and caused 463 deaths in 23 countries between 2003 and April 2024, according to the World Health Organization (WHO). On April 1, 2024, the CDC reported a case of highly pathogenic avian influenza A H5N1 in a farmworker in Texas during a multistate outbreak of avian influenza in dairy cows. Two more cases were subsequently reported in farmworkers in Michigan.
The CDC analyzed sera (blood) collected from people of all ages in all 10 Health & Human Services regions during the 2022-2023 and 2021-2022 flu seasons. These samples were challenged with H5N1 virus to see whether there was an antibody reaction. Data from this study suggest that there is extremely low to no population immunity to clade 2.3.4.4b A (H5N1) viruses in the U.S. Antibody levels remained low regardless of whether or not the participants received a seasonal flu vaccination, meaning that seasonal flu vaccination did not produce antibodies to H5N1 viruses.
Cocrystal Pharma determined the high resolution X-ray crystal structure of the recent avian influenza A (H5N1) PB2 protein and confirmed activity of CC-42344 in vitro (NIH GeneBank ID:influenza A/Texas/37/2024(H5N1). The crystal structure of the avian influenza A (H5N1) PB2 protein showed new mutations located outside the PB2 active site. Subsequent studies showed that CC-42344 binds to the active site of the avian influenza A (H5N1) PB2 protein as previously demonstrated with the pandemic and seasonal influenza A PB2. Preliminary in vitro assays confirmed that CC-42344 exhibits high potency against the avian influenza A (H5N1) PB2 protein.
About CC-42344 CC-42344 is Cocrystal’s novel, broad-spectrum, antiviral investigational candidate for the treatment of pandemic and seasonal influenza A. CC-42344 inhibits the first step in influenza A’s viral replication by binding to a highly conserved PB2 site of the influenza polymerase complex that is essential to replication and was discovered using Cocrystal’s proprietary structure-based drug discovery platform technology.
Cocrystal is conducting a Phase 2a human challenge study in the United Kingdom to evaluate safety, viral and clinical measures of oral CC-42344 in healthy volunteers who are challenged with influenza A. CC-42344 was advanced into Phase 2a testing following favorable safety and tolerability results reported in a Phase 1 study in healthy volunteers conducted in Australia. In vitro testing showed CC-42344’s excellent antiviral activity against influenza A strains, including pandemic and seasonal strains, as well as against strains resistant to Tamiflu® and Xofluza®, while also demonstrating favorable pharmacokinetic and safety profiles.
About Cocrystal Pharma, Inc. Cocrystal Pharma, Inc. is a clinical-stage biotechnology company discovering and developing novel antiviral therapeutics that target the replication process of influenza viruses, coronaviruses (including SARS-CoV-2), noroviruses and hepatitis C viruses. Cocrystal employs unique structure-based technologies and Nobel Prize-winning expertise to create first- and best-in-class antiviral drugs. For further information about Cocrystal, please visit www.cocrystalpharma.com.
Cautionary Note Regarding Forward-Looking Statements This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the potential efficacy of CC-42344 against the avian influenza A H5N1 virus, the expected timing and results of the Phase 2a trial for CC-42344 for the oral treatment of influenza A in 2024, and the potential market for such product candidate. The words “believe,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “could,” “target,” “potential,” “is likely,” “will,” “expect” and similar expressions, as they relate to us, are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events. Some or all of the events anticipated by these forward-looking statements may not occur. Important factors that could cause actual results to differ from those in the forward-looking statements include, but are not limited to, risks relating to our ability to obtain regulatory authority for and proceed with clinical trials including recruiting volunteers and procuring materials for such studies by our clinical research organizations and vendors, the results of such studies, our and our collaboration partners’ technology and software performing as expected, general risks arising from clinical studies, receipt of regulatory approvals, regulatory changes, and potential development of effective treatments and/or vaccines by competitors, including as part of the programs financed by the U.S. government, potential mutations in a virus we are targeting that may result in variants that are resistant to a product candidate we develop. Further information on our risk factors is contained in our filings with the SEC, including our Annual Report on Form 10-K for the year ended December 31, 2023. Any forward-looking statement made by us herein speaks only as of the date on which it is made. Factors or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of them. We undertake no obligation to publicly update any forward-looking statement, whether as a result of new information, future developments or otherwise, except as may be required by law.
MALVERN, Pa., June 20, 2024 (GLOBE NEWSWIRE) — Ocugen, Inc. (“Ocugen” or the “Company”) (NASDAQ: OCGN), a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines, today announced that the first patient has been dosed in its Phase 3 liMeliGhT clinical trial for OCU400—a modifier gene therapy product candidate being developed for retinitis pigmentosa (RP).
“Each clinical milestone achieved by OCU400 brings us closer to providing a one-time treatment for life to patients living with RP,” said Dr. Shankar Musunuri, Chairman, CEO and Co-founder of Ocugen. “Dosing the first patient is especially significant and makes our dedication to serving RP patients—300,000 in the U.S. and Europe and 1.6 million worldwide—more tangible.”
The Phase 3 liMeliGhT clinical trial was informed by positive Phase 1/2 OCU400 data that suggests positive trends in Best-Corrected Visual Acuity (BCVA) and Multi-Luminance Mobility Testing (MLMT), and Low-Luminance Visual Acuity (LLVA) among treated eyes. 89% (16/18) of RP subjects demonstrated preservation or improvement in the treated eye either on BCVA or LLVA or MLMT scores from baseline. 80% (8/10) of RHO mutation subjects experienced either preservation or improvement in MLMT scores from baseline. 78% (14/18) of subjects demonstrated preservation or improvement in treated eyes in MLMT scores from baseline.
The Phase 3 study—with the duration of one year—will have a sample size of 150 participants—one arm of 75 participants with RHO gene mutations and the other arm with 75 participants that are gene agnostic. In each arm, participants will be randomized 2:1 to the treatment group (2.5 x 1010 vg/eye of OCU400) and untreated control group, respectively. Patients eight years of age and older, with early through late-stage advancement of RP, are being recruited to participate in the liMeliGhT study.
Luminance Dependent Navigation Assessment (LDNA)—a more sensitive and specific measurement of function than MLMT used in previous Phase 3 clinical trials—is the primary endpoint for the study. The Phase 3 liMeliGhT study will focus on the proportion of responders, in treated and untreated groups, achieving an improvement of at least 2 Lux levels from baseline in the study eyes.
“Patients with RP associated with mutations in multiple genes currently have no therapeutic options. As a retinal surgeon, I am encouraged by the therapeutic potential of OCU400 to provide long-term benefit to RP patients,” said Lejla Vajzovic, MD, FASRS, Director, Duke Surgical Vitreoretinal Fellowship Program, Associate Professor of Ophthalmology with Tenure Adult and Pediatric Vitreoretinal Surgery and Disease, Duke University Eye Center, and Retina Scientific Advisory Board Chair of Ocugen. “OCU400 is a novel modifier gene therapy approach that could initiate a paradigm shift in the treatment of RP and to field of ophthalmology.”
“The current OCU400 Phase 3 study is very exciting and gives hope for thousands of individuals with RP,” said Benjamin Bakall, MD, PhD, Director of Clinical Research at Associated Retina Consultants (ARC) and Clinical Assistant Professor at University of Arizona, College of Medicine – Phoenix. “I am encouraged that we may have a potential treatment option to preserve or improve the vision in RP patients regardless of gene mutation, and very pleased that the first patient dosing in the Phase 3 liMeliGhT clinical trial was performed at ARC.”
“We are grateful for our continued collaboration with Dr. Bakall and the team at ARC,” said Dr. Huma Qamar, Chief Medical Officer of Ocugen. “We are excited to expand our enrollment to include more centers and patients representing a diverse array of RP gene mutations, which will be a validation of this novel gene therapy platform. We will provide updates as our progress continues.”
Ocugen previously announced that OCU400 has received orphan drug and RMAT designations from the FDA and that the EMA provided acceptability of the U.S.-based trial for submission of a Marketing Authorization Application (MAA). With the first dosing of the Phase 3 clinical trial, OCU400 remains on track for the 2026 BLA and MAA approval targets.
About OCU400 OCU400 is the Company’s gene-agnostic modifier gene therapy product based on nuclear hormone receptor (NHR) gene, NR2E3. NR2E3 regulates diverse physiological functions within the retina—such as photoreceptor development and maintenance, metabolism, phototransduction, inflammation and cell survival networks. Through its drive functionality, OCU400 resets altered/affected cellular gene networks and establishes homeostasis—a state of balance, which has the potential to improve retinal health and function in patients with RP.
AboutModifierGeneTherapy Modifier gene therapy is designed to fulfill unmet medical needs related to retinal diseases, including IRDs, such as RP, LCA and Stargardt disease, as well as multifactorial diseases like dry age-related macular degeneration (dAMD). Our modifier gene therapy platform is based on the use of NHRs, master gene regulators, which have the potential to restore homeostasis — the basic biological processes in the retina. Unlike single-gene replacement therapies, which only target one genetic mutation, we believe that our modifier gene therapy platform, through its use of NHRs, represents a novel approach that has the potential to address multiple retinal diseases caused by mutations in multiple genes with one product, and to address complex diseases that are potentially caused by imbalances in multiple gene networks. Currently, Ocugen has three modifier gene therapy programs in the clinic: OCU400, OCU410, and OCU410ST. In addition to the OCU400 Phase 3 liMeliGhT clinical trial, the OCU410 Phase 1/2 ArMaDa clinical trial for geographic atrophy (GA) secondary to dAMD and the OCU410ST Phase 1/2 GARDian clinical trial for Stargardt disease are currently underway. GA affects approximately two to three million people in the U.S. and EU combined and Stargardt disease affects nearly 100,000 people in the U.S. and EU combined.
About Ocugen, Inc. Ocugen, Inc. is a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines that improve health and offer hope for patients across the globe. We are making an impact on patient’s lives through courageous innovation—forging new scientific paths that harness our unique intellectual and human capital. Our breakthrough modifier gene therapy platform has the potential to treat multiple retinal diseases with a single product, and we are advancing research in infectious diseases to support public health and orthopedic diseases to address unmet medical needs. Discover more at www.ocugen.com and follow us on X and LinkedIn.
Cautionary Note on Forward-Looking Statements This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995, including, but not limited to, statements regarding qualitative assessments of available data, potential benefits, expectations for ongoing clinical trials, anticipated regulatory filings and anticipated development timelines, which are subject to risks and uncertainties. We may, in some cases, use terms such as “predicts,” “believes,” “potential,” “proposed,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Such statements are subject to numerous important factors, risks, and uncertainties that may cause actual events or results to differ materially from our current expectations, including, but not limited to, the risks that preliminary, interim and top-line clinical trial results may not be indicative of, and may differ from, final clinical data; that unfavorable new clinical trial data may emerge in ongoing clinical trials or through further analyses of existing clinical trial data; that earlier non-clinical and clinical data and testing of may not be predictive of the results or success of later clinical trials; and that that clinical trial data are subject to differing interpretations and assessments, including by regulatory authorities. These and other risks and uncertainties are more fully described in our periodic filings with the Securities and Exchange Commission (SEC), including the risk factors described in the section entitled “Risk Factors” in the quarterly and annual reports that we file with the SEC. Any forward-looking statements that we make in this press release speak only as of the date of this press release. Except as required by law, we assume no obligation to update forward-looking statements contained in this press release whether as a result of new information, future events, or otherwise, after the date of this press release.
10,000-participant randomized Phase 2b study will evaluate and compare GeoVax’s multi-antigen, vaccine candidate (GEO-CM04S1) to an approved vaccine against COVID-19 under BARDA’s Clinical Studies Network
Project NextGen is a $5 billion initiative by the U.S. Department of Health and Human Services to develop innovative vaccines and therapeutics providing broader and more durable protection against current and future COVID-19 viral strains
Atlanta, GA, June 18, 2024 – GeoVax Labs, Inc. (Nasdaq: GOVX), a biotechnology company developing immunotherapies and vaccines against cancers and infectious diseases, today announced that it received an award through the Rapid Response Partnership Vehicle (RRPV) to advance development of GEO-CM04S1, GeoVax’s dual-antigen next-generation COVID-19 vaccine, in a Phase 2b clinical trial. The RRPV is a Consortium funded by the Biomedical Advanced Research and Development Authority (BARDA), part of the Administration for Strategic Preparedness and Response (ASPR) in the U.S. Department of Health and Human Services (HHS).
Under the agreement, GeoVax will sponsor a 10,000-participant, randomized, Phase 2b double-blinded study to compare the efficacy, safety, and immunogenicity of GEO-CM04S1 with a U.S. Food and Drug Administration (FDA)-approved mRNA COVID-19 vaccine. Preparations for the study are underway, and execution of the study will be fully funded by BARDA under its Clinical Studies Network.
The direct award to GeoVax of approximately $24.3 million, which may increase to as much as $45 million, will fund the manufacturing of clinical materials and support for the Phase 2b clinical trial, including regulatory activities. BARDA has made separate awards through its Clinical Studies Network to support execution of the study. That funding will represent approximately $343M from the Project NextGen program for a CRO to execute the clinical trial using GeoVax’s vaccine.
“We are honored and proud to receive this award from BARDA to advance our next-generation vaccine against COVID-19. This contract not only provides the vital resources for advancing the development of GEO-CM04S1, but it also advances our MVA platform in infectious diseases,” said David Dodd, Chairman & CEO of GeoVax.
Mr. Dodd continued, “First-generation COVID-19 vaccines were beneficial during the peak of the pandemic but are limited in breadth and durability of clinical protection, requiring frequent updates. Now that COVID-19 is in an endemic stage, many people continue to have their everyday lives impacted in meaningful ways. Furthermore, there are an estimated 23 million adults in the U.S. with immunocompromised conditions who are less likely to have an adequate response to current vaccines and are more likely to potentially experience severe COVID-19 symptoms, hospitalization and the risk of death, even after vaccination. GEO-CM04S1 was designed to address these limitations by inducing durable neutralizing antibody and T-cell-based immunity against current and future SARS-CoV-2 variants. Our vaccine has continued to demonstrate induction of potent immune responses with potential to drive broad and durable clinical protection, and we eagerly anticipate commencing the Phase 2b study to further demonstrate the value and advantages of our technology.”
Funding for this award is provided under Project NextGen, a $5 billion initiative by HHS to advance a pipeline of new, innovative vaccines and therapeutics providing broader and more durable protection for COVID-19 than the first generation COVID vaccines and medicines. BARDA is supporting the development of new vaccines and therapeutics to better address the waning immunity and resistance to current and future SARS-CoV2 viral strains. GeoVax’s vaccine candidate provides many of the features identified by BARDA including broader protection among variants of concern (VOC) and a longer duration of protection.
This project is being funded with federal funds from the Department of Health and Human Services; Administration for Strategic Preparedness and Response (ASPR); Biomedical Advanced Research and Development Authority (BARDA), under Other Transaction (OT) number: 75A50123D00005.
About GEO-CM04S1
GEO-CM04S1 is based on GeoVax’s MVA viral vector platform, which supports the presentation of multiple vaccine antigens to the immune system in a single dose. GEO-CM04S1 encodes for both the spike (S) and nucleocapsid (N) antigens of SARS-CoV-2 and is specifically designed to induce both antibody and T-cell responses to those parts of the virus less likely to mutate over time. The more broadly functional engagement of the immune system is designed to protect against severe disease caused by continually emerging variants of COVID-19. Vaccines of this format should not require frequent and repeated modification or updating.
GEO-CM04S1 is currently being evaluated in three ongoing Phase 2 clinical trials:
As a primary vaccine in immunocompromised patients (with hematologic cancers receiving cell transplants or CAR-T therapy). ClinicalTrials.gov Identifier: NCT04977024. A recent presentation of unpublished data from the open-label portion of the trial indicates that GEO-CM04S1 is highly immunogenic in these patients, inducing both antibody responses, including neutralizing antibodies, and T-cell responses.
As a booster vaccine in immunocompromised patients with chronic lymphocytic leukemia (CLL), a recognized high-risk group for whom current mRNA vaccines and monoclonal antibody (MAb) therapies appear inadequate relative to providing protective immunity. ClinicalTrials.gov Identifier: NCT05672355.
As a booster vaccine for healthy adults who have previously received the Pfizer or Moderna mRNA vaccine. gov Identifier: NCT04639466.
About GeoVax
GeoVax Labs, Inc. is a clinical-stage biotechnology company developing novel therapies and vaccines for solid tumor cancers and many of the world’s most threatening infectious diseases. The company’s lead program in oncology is a novel oncolytic solid tumor gene-directed therapy, Gedeptin®, which recently completed enrollment in a multicenter Phase 1/2 clinical trial for advanced head and neck cancers. GeoVax’s lead infectious disease candidate is GEO-CM04S1, a next-generation COVID-19 vaccine targeting high-risk immunocompromised patient populations. Currently in three Phase 2 clinical trials, GEO-CM04S1 is being evaluated as a primary vaccine for immunocompromised patients such as those suffering from hematologic cancers and other patient populations for whom the current authorized COVID-19 vaccines are insufficient, and as a booster vaccine in patients with chronic lymphocytic leukemia (CLL). In addition, GEO-CM04S1 is in a Phase 2 clinical trial evaluating the vaccine as a more robust, durable COVID-19 booster among healthy patients who previously received the mRNA vaccines. GeoVax has a leadership team who have driven significant value creation across multiple life science companies over the past several decades. For more information, visit our website: www.geovax.com.
Forward-Looking Statements
This release contains forward-looking statements regarding GeoVax’s business plans. The words “believe,” “look forward to,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “could,” “target,” “potential,” “is likely,” “will,” “expect” and similar expressions, as they relate to us, are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. Actual results may differ materially from those included in these statements due to a variety of factors, including whether: GeoVax is able to obtain acceptable results from ongoing or future clinical trials of its investigational products, GeoVax’s immuno-oncology products and preventative vaccines can provoke the desired responses, and those products or vaccines can be used effectively, GeoVax’s viral vector technology adequately amplifies immune responses to cancer antigens, GeoVax can develop and manufacture its immuno-oncology products and preventative vaccines with the desired characteristics in a timely manner, GeoVax’s immuno-oncology products and preventative vaccines will be safe for human use, GeoVax’s vaccines will effectively prevent targeted infections in humans, GeoVax’s immuno-oncology products and preventative vaccines will receive regulatory approvals necessary to be licensed and marketed, GeoVax raises required capital to complete development, there is development of competitive products that may be more effective or easier to use than GeoVax’s products, GeoVax will be able to enter into favorable manufacturing and distribution agreements, and other factors, over which GeoVax has no control.
Further information on our risk factors is contained in our periodic reports on Form 10-Q and Form 10-K that we have filed and will file with the SEC. Any forward-looking statement made by us herein speaks only as of the date on which it is made. Factors or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of them. We undertake no obligation to publicly update any forward-looking statement, whether as a result of new information, future developments or otherwise, except as may be required by law.
Gap in non-oral prescriptions relative to AHS guidelines represents an opportunity to increase awareness of Tonix’s two FDA-approved non-oral treatments for acute migraine
Zembrace® SymTouch® (sumatriptan injection) and Tosymra® (sumatriptan nasal spray) are indicated for the treatment of acute migraine in adults
CHATHAM, N.J., June 18, 2024 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP) (Tonix or the Company), a fully-integrated biopharmaceutical company with marketed products and a pipeline of development candidates, presented data from a poster presentation at the 66th Annual Scientific Meeting of the American Headache Society (AHS), held June 13-16, 2024. A copy of the Company’s poster presentation is available under the Scientific Presentations tab of the Tonix website at www.tonixpharma.com.
In the poster presentation titled, “American Headache Society Consensus Statement and Other Recommendations: How Many Practitioners Comply With the Recommendations?,” a retrospective review of real-world data compares real world usage of non-oral migraine products with the most recent AHS consensus statement. The data reaffirms several past recommendations from the AHS and stresses the need for customizing treatment of migraine headaches to the needs of patients, as well as using the most appropriate route of administration for any given acute attack based on the clinical presentation. So far, real world data show that compliance with the guidelines and the consensus statement have yet to be achieved but has the potential to be increased. Specifically, the data show the use of non-oral drugs for treating an acute migraine attack was only 7% in 2012 and has decreased to below 4% in 2023, when the potential need for such drugs is anticipated to be a significant percentage of patients based on epidemiological data.
“We believe personalizing therapy for migraine is the future and it is hoped that non-oral medicines will address some of the persistent dissatisfaction patients experience with their migraine treatments,” said Seth Lederman, M.D., Chief Executive Officer of Tonix Pharmaceuticals. “We hope that educating prescribers about the importance of customized treatments will lead to enhanced satisfaction and improve the quality of life of migraine patients. This represents an opportunity for growth in non-oral first-line therapeutics such Tonix’s Zembrace SymTouch and Tosymra.”
“Inconsistent and incomplete response to traditional or emerging oral acute migraine medications continue to be a burden for individuals living with migraines. We believe that both Zembrace SymTouch and Tosymra, due to their rapid-onset and route of administration are well suited to address this unmet need,” said James Hunter, Executive Vice President of Commercial Operations at Tonix Pharmaceuticals and President of Tonix Medicines.
Zembrace SymTouch is the only actively promoted brand of sumatriptan autoinjector in the U.S. (other sumatriptan autoinjector products on the market are Imitrex® and generics to Imitrex®). It has a unique low dose and has demonstrated onset of migraine pain relief in as few as 10 minutes (17% of patients vs. 5% for placebo)1. Zembrace SymTouch also demonstrated migraine pain freedom for 46% of patients (vs 27% for placebo) at 2 hours in a single-attack, double-blind study (N=230)2. Tosymra employs Intravail® permeation enhancer technology and is pharmacokinetically equivalent to 4 mg subcutaneous sumatriptan.3,4, Tosymra delivers migraine pain relief in as little as 10 minutes with just one spray for some patients (13% vs. 5% for placebo).1,4,5
About Migraine
Nearly 40 million people in the United States suffer from migraine6 and it has been recognized as the second leading cause of disability in the world7. Migraine is characterized by debilitating attacks lasting four to 72 hours with multiple symptoms, including pulsating headaches of moderate to severe pain intensity often associated with nausea or vomiting, and/or sensitivity to sound (phonophobia) and sensitivity to light (photophobia)8.
1Mathew NT, et al. Dose ranging efficacy and safety of subcutaneous sumatriptan in the acute treatment of migraine. US Sumatriptan Research Group. Arch Neurol. 1992;49(12):1271-1276.
2Landy, S. et al. Efficacy and safety of DFN-11 (sumatriptan injection, 3 mg) in adults with episodic migraine: a multicenter, randomized, double-blind, placebo-controlled study. J Headache Pain. 19, 69 (2018).
3Brand-Schieber E, Munjal S, Kumar R, et al. Human factors validation study of 3 mg sumatriptan autoinjector, for migraine patients. Med Devices (Auckl). 2016;9:131-137.
4Tosymra [package insert]. Maple Grove, MN: Upsher-Smith Laboratories, LLC: Feb 2021.
5Wendt J, et al. A randomized, double-blind, placebo-controlled trial of the efficacy and tolerability of a 4-mg dose of subcutaneous sumatriptan for the treatment of acute migraine attacks in adults. Clinical Therapeutics. 2006;28(4):517-526.
6IQVIA 2022 retail sales from the National Sales Perspectives (NSP) audit within the SMART database estimates Zembrace sales of ~$19.6 M and Tosymra sales of ~$3.5 M
7GBD 2016 Headache Collaborators. Global, regional, and national burden of migraine and tension-type headache, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2018;17(11):954-976.
8Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition. Cephalalgia. 2018;38(1):1–211.
Tonix Pharmaceuticals Holding Corp.*
Tonix is a fully-integrated biopharmaceutical company focused on developing, licensing and commercializing therapeutics to treat and prevent human disease and alleviate suffering. Tonix’s development portfolio is focused on central nervous system (CNS) disorders. Tonix’s priority is to submit a New Drug Application (NDA) to the FDA in the second half of 2024 for Tonmya1, a product candidate for which two statistically significant Phase 3 studies have been completed for the management of fibromyalgia. TNX-102 SL is also being developed to treat acute stress reaction as well as fibromyalgia-type Long COVID. Tonix’s CNS portfolio includes TNX-1300 (cocaine esterase), a biologic designed to treat cocaine intoxication that has FDA Breakthrough Therapy designation and is in Phase 2 development supported by a grant from the National institute of Drug Abuse. Tonix’s immunology development portfolio consists of biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases. Tonix also has product candidates in development in the areas of rare disease and infectious disease. Tonix Medicines, our commercial subsidiary, markets Zembrace® SymTouch® (sumatriptan injection) 3 mg and Tosymra® (sumatriptan nasal spray) 10 mg for the treatment of acute migraine with or without aura in adults.
*Tonix’s product development candidates are investigational new drugs or biologics and have not been approved for any indication.
1Tonmya™ is conditionally accepted by the U.S. Food and Drug Administration (FDA) as the tradename for TNX-102 SL for the management of fibromyalgia. Tonmya has not been approved for any indication.
Zembrace SymTouch and Tosymra are registered trademarks of Tonix Medicines. All other marks are property of their respective owners.
This press release and further information about Tonix can be found at www.tonixpharma.com.
Forward Looking Statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; risks related to the failure to successfully market any of our products; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2023, as filed with the Securities and Exchange Commission (the “SEC”) on April 1, 2024, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
Zembrace® SymTouch® (sumatriptan Injection): IMPORTANT SAFETY INFORMATION
Zembrace SymTouch (Zembrace) can cause serious side effects, including heart attack and other heart problems, which may lead to death. Stop use and get emergency help if you have any signs of a heart attack:
discomfort in the center of your chest that lasts for more than a few minutes or goes away and comes back
severe tightness, pain, pressure, or heaviness in your chest, throat, neck, or jaw
pain or discomfort in your arms, back, neck, jaw or stomach
shortness of breath with or without chest discomfort
breaking out in a cold sweat
nausea or vomiting
feeling lightheaded
Zembrace is not for people with risk factors for heart disease (high blood pressure or cholesterol, smoking, overweight, diabetes, family history of heart disease) unless a heart exam shows no problem.
Do not use Zembrace if you have:
history of heart problems
narrowing of blood vessels to your legs, arms, stomach, or kidney (peripheral vascular disease)
uncontrolled high blood pressure
hemiplegic or basilar migraines. If you are not sure if you have these, ask your provider.
had a stroke, transient ischemic attacks (TIAs), or problems with blood circulation
severe liver problems
taken any of the following medicines in the last 24 hours: almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, ergotamines, dihydroergotamine.
are taking certain antidepressants, known as monoamine oxidase (MAO)-A inhibitors or it has been 2 weeks or less since you stopped taking a MAO-A inhibitor. Ask your provider for a list of these medicines if you are not sure.
an allergy to sumatriptan or any of the components of Zembrace
Tell your provider about all of your medical conditions and medicines you take, including vitamins and supplements.
Zembrace can cause dizziness, weakness, or drowsiness. If so, do not drive a car, use machinery, or do anything where you need to be alert.
Zembrace may cause serious side effects including:
changes in color or sensation in your fingers and toes
sudden or severe stomach pain, stomach pain after meals, weight loss, nausea or vomiting, constipation or diarrhea, bloody diarrhea, fever
cramping and pain in your legs or hips; feeling of heaviness or tightness in your leg muscles; burning or aching pain in your feet or toes while resting; numbness, tingling, or weakness in your legs; cold feeling or color changes in one or both legs or feet
increased blood pressure including a sudden severe increase even if you have no history of high blood pressure
medication overuse headaches from using migraine medicine for 10 or more days each month. If your headaches get worse, call your provider.
serotonin syndrome, a rare but serious problem that can happen in people using Zembrace, especially when used with anti-depressant medicines called SSRIs or SNRIs. Call your provider right away if you have: mental changes such as seeing things that are not there (hallucinations), agitation, or coma; fast heartbeat; changes in blood pressure; high body temperature; tight muscles; or trouble walking.
hives (itchy bumps); swelling of your tongue, mouth, or throat
seizures even in people who have never had seizures before
The most common side effects of Zembrace include: pain and redness at injection site; tingling or numbness in your fingers or toes; dizziness; warm, hot, burning feeling to your face (flushing); discomfort or stiffness in your neck; feeling weak, drowsy, or tired.
Tell your provider if you have any side effect that bothers you or does not go away. These are not all the possible side effects of Zembrace. For more information, ask your provider.
This is the most important information to know about Zembrace but is not comprehensive. For more information, talk to your provider and read the Patient Information and Instructions for Use. You can also visit www.tonixpharma.com or call 1-888-650-3789.
You are encouraged to report adverse effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088.
INDICATION AND USAGE
Zembrace is a prescription medicine used to treat acute migraine headaches with or without aura in adults who have been diagnosed with migraine.
Zembrace is not used to prevent migraines. It is not known if it is safe and effective in children under 18 years of age.
Tosymra can cause serious side effects, including heart attack and other heart problems, which may lead to death. Stop Tosymra and get emergency medical help if you have any signs of heart attack:
discomfort in the center of your chest that lasts for more than a few minutes or goes away and comes back
severe tightness, pain, pressure, or heaviness in your chest, throat, neck, or jaw
pain or discomfort in your arms, back, neck, jaw, or stomach
shortness of breath with or without chest discomfort
breaking out in a cold sweat
nausea or vomiting
feeling lightheaded
Tosymra is not for people with risk factors for heart disease (high blood pressure or cholesterol, smoking, overweight, diabetes, family history of heart disease) unless a heart exam is done and shows no problem.
Do not use Tosymra if you have:
history of heart problems
narrowing of blood vessels to your legs, arms, stomach, or kidney (peripheral vascular disease)
uncontrolled high blood pressure
severe liver problems
hemiplegic or basilar migraines. If you are not sure if you have these, ask your healthcare provider.
had a stroke, transient ischemic attacks (TIAs), or problems with blood circulation
taken any of the following medicines in the last 24 hours: almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, ergotamines, or dihydroergotamine. Ask your provider if you are not sure if your medicine is listed above.
are taking certain antidepressants, known as monoamine oxidase (MAO)-A inhibitors or it has been 2 weeks or less since you stopped taking a MAO-A inhibitor. Ask your provider for a list of these medicines if you are not sure.
an allergy to sumatriptan or any ingredient in Tosymra
Tell your provider about all of your medical conditions and medicines you take, including vitamins and supplements.
Tosymra can cause dizziness, weakness, or drowsiness. If so, do not drive a car, use machinery, or do anything where you need to be alert.
Tosymra may cause serious side effects including:
changes in color or sensation in your fingers and toes
sudden or severe stomach pain, stomach pain after meals, weight loss, nausea or vomiting, constipation or diarrhea, bloody diarrhea, fever
cramping and pain in your legs or hips, feeling of heaviness or tightness in your leg muscles, burning or aching pain in your feet or toes while resting, numbness, tingling, or weakness in your legs, cold feeling or color changes in one or both legs or feet
increased blood pressure including a sudden severe increase even if you have no history of high blood pressure
medication overuse headaches from using migraine medicine for 10 or more days each month. If your headaches get worse, call your provider.
serotonin syndrome, a rare but serious problem that can happen in people using Tosymra, especially when used with anti-depressant medicines called SSRIs or SNRIs. Call your provider right away if you have: mental changes such as seeing things that are not there (hallucinations), agitation, or coma; fast heartbeat; changes in blood pressure; high body temperature; tight muscles; or trouble walking.
hives (itchy bumps); swelling of your tongue, mouth, or throat
seizures even in people who have never had seizures before
The most common side effects of Tosymra include: tingling, dizziness, feeling warm or hot, burning feeling, feeling of heaviness, feeling of pressure, flushing, feeling of tightness, numbness, application site (nasal) reactions, abnormal taste, and throat irritation.
Tell your provider if you have any side effect that bothers you or does not go away. These are not all the possible side effects of Tosymra. For more information, ask your provider.
This is the most important information to know about Tosymra but is not comprehensive. For more information, talk to your provider and read the Patient Information and Instructions for Use. You can also visit www.tonixpharma.com or call 1-888-650-3789.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088.
INDICATION AND USAGE Tosymra is a prescription medicine used to treat acute migraine headaches with or without aura in adults.
Tosymra is not used to treat other types of headaches such as hemiplegic or basilar migraines or cluster headaches.
Tosymra is not used to prevent migraines. It is not known if Tosymra is safe and effective in children under 18 years of age.