MALVERN, Pa., Aug. 08, 2025 (GLOBE NEWSWIRE) — Ocugen, Inc. (Ocugen or the Company) (NASDAQ: OCGN), a pioneering biotechnology leader in gene therapies for blindness diseases, today announced that it has entered into a securities purchase agreement with Janus Henderson Investors, a global asset management firm, to purchase 20,000,000 shares of common stock and warrants to purchase up to an aggregate of 20,000,000 shares of common stock at a purchase price of $1.00 per share (closing price on August 7, 2025) and accompanying warrant in a registered direct offering. The warrants have an exercise price of $1.50 per share, are exercisable immediately upon issuance, and will expire two years following the date of issuance. The warrants are callable by the Company when the VWAP of the Company’s common stock exceeds $2.50 per share for at least five of a trailing 30 trading day period.
Noble Capital Markets, Inc. acting as the sole placement agent in connection with the offering.
The gross proceeds to the Company are expected to be approximately $20 million before deducting the placement agent fees and other estimated offering expenses. The Company may receive up to $30 million of additional gross proceeds if the warrants are exercised in full. The offering is expected to close on or about August 11, 2025, subject to the satisfaction of customary closing conditions. The offering is being made pursuant to an effective shelf registration statement on Form S-3 (File No. 333-278774) previously filed with the U.S. Securities and Exchange Commission (“SEC”), which was declared effective on May 1, 2024. The offering is made only by means of a prospectus forming a part of the effective registration statement relating to the offering. A prospectus supplement relating to the shares of common stock and warrants will be filed by the Company with the SEC. When available, copies of the prospectus supplement relating to the registered direct offering, together with the accompanying prospectus, can be obtained at the SEC’s website at www.sec.gov or from Noble Capital Markets, Inc., 150 East Palmetto Park Rd., Suite 110 Boca Raton, FL 33432.
This press release shall not constitute an offer to sell or a solicitation of an offer to buy any of the securities described herein, nor shall there be any sale of these securities in any state or other jurisdiction in which such offer, solicitation or sale would be unlawful prior to the registration or qualification under the securities laws of any such state or other jurisdiction.
AboutOcugen,Inc.
Ocugen, Inc. is a pioneering biotechnology leader in gene therapies for blindness diseases. Our breakthrough modifier gene therapy platform has the potential to address significant unmet medical need for large patient populations through our gene-agnostic approach. Unlike traditional gene therapies and gene editing, Ocugen’s modifier gene therapies address the entire disease—complex diseases that are potentially caused by imbalances in multiple gene networks. Currently we have programs in development for inherited retinal diseases and blindness diseases affecting millions across the globe, including retinitis pigmentosa, Stargardt disease, and geographic atrophy—late stage dry age-related macular degeneration. Discover more at www.ocugen.com and follow us on X and LinkedIn.
This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995, which are subject to risks and uncertainties. We may, in some cases, use terms such as “predicts,” “believes,” “potential,” “proposed,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Such statements are subject to numerous important factors, risks, and uncertainties that may cause actual events or results to differ materially from our current expectations. Further, certain forward-looking statements are based on assumptions as to future events that may not prove to be accurate, including the satisfaction of customary closing conditions related to the offering, completion of the offering, whether the warrants will be exercised and various other factors. These and other risks and uncertainties are more fully described in our periodic filings with the Securities and Exchange Commission (SEC), including the risk factors described in the section entitled “Risk Factors” in the quarterly and annual reports that we file with the SEC. Any forward-looking statements that we make in this press release speak only as of the date of this press release. Except as required by law, we assume no obligation to update forward-looking statements contained in this press release whether as a result of new information, future events, or otherwise, after the date of this press release.
DLH delivers improved health and readiness solutions for federal programs through research, development, and innovative care processes. The Company’s experts in public health, performance evaluation, and health operations solve the complex problems faced by civilian and military customers alike, leveraging digital transformation, artificial intelligence, advanced analytics, cloud-based applications, telehealth systems, and more. With over 2,300 employees dedicated to the idea that “Your Mission is Our Passion,” DLH brings a unique combination of government sector experience, proven methodology, and unwavering commitment to public health to improve the lives of millions. For more information, visit www.DLHcorp.com.
Joe Gomes, CFA, Managing Director, Equity Research Analyst, Generalist , Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
When, Not If. We continue to believe it is a matter of when, not if, DLH begins to capitalize on the large opportunity set for its mission critical skill set. Current disruptions in Federal government contracting will pass, and DLH’s capabilities, in areas such as digital transformation, cybersecurity, and addressing critical public health issues, align well with the government’s goals.
Still Accumulating. Mink Brook Asset Management continues to accumulate DLHC shares, including 5,900 shares at the end of last week. Mink Brook now owns 2,389,350 DLHC shares, representing 16.6% of the outstanding common, up from 2,164,058 shares at the end of May.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
FDA grants meeting for AVERSA™ FENTANYL (abuse deterrent transdermal system) to provide feedback on the Chemistry, Manufacturing, and Controls plans for the product through commercialization.
Nutriband is partnering with Kindeva to develop AVERSA™ FENTANYL which combines Nutriband’s AVERSA™ abuse-deterrent technology with Kindeva’s FDA-approved fentanyl patch.
ORLANDO, Fla., Aug. 08, 2025 (GLOBE NEWSWIRE) — Nutriband Inc. (NASDAQ:NTRB)(NASDAQ:NTRBW), a company engaged in the development of prescription transdermal pharmaceutical products, today announced that the United States Food and Drug Administration (US FDA) has granted a Type C Meeting for its lead product, AVERSA™ FENTANYL (abuse deterrent fentanyl transdermal system). The purpose of the meeting is to specifically provide feedback on the Chemistry, Manufacturing, and Controls (CMC) plans for the product from submission of an Investigational New Drug Application (IND) through approval of a 505(b)(2) New Drug Application (NDA) and subsequent commercialization.
The meeting is scheduled as a virtual face-to-face meeting to be held on September 18, 2025 with the Division of Anesthesiology, Addiction Medicine, and Pain Medicine (DAAP) in the Office of Neuroscience (ON), Center for Drug Evaluation and Research (CDER).
Nutriband is partnering with Kindeva to develop AVERSA™ FENTANYL which combines Nutriband’s AVERSA™ abuse-deterrent technology with Kindeva’s FDA-approved fentanyl patch.
Nutriband’s AVERSA™ abuse-deterrent technology can be utilized to incorporate aversive agents into transdermal patches to prevent the abuse, diversion, misuse, and accidental exposure of drugs with abuse potential including opioids and stimulants. The AVERSA™ abuse-deterrent technology has the potential to improve the safety profile of transdermal drugs susceptible to abuse, such as fentanyl, while making sure that these drugs remain accessible to those patients who really need them.
AVERSA FENTANYL has the potential to be the world’s first abuse-deterrent opioid patch designed to deter the abuse and misuse and reduce the risk of accidental exposure of transdermal fentanyl patches. AVERSA FENTANYL has the potential to reach peak annual US sales of $80 million to $200 million.1 While initially concentrating on the US market, the unmet medical need for adequate pain management is a global problem, and our goal is to make AVERSA FENTANYL available in all major medical markets in the world.
The AVERSA™ abuse deterrent technology is protected by a broad international intellectual property portfolio with patents issued in 46 countries including the United States, Europe, Japan, Korea, Russia, China, Canada, Mexico, and Australia.
1 Health Advances Aversa Fentanyl market analysis report 2022
About AVERSA™ Abuse-Deterrent Transdermal Technology
Nutriband’s AVERSA™ abuse-deterrent transdermal technology incorporates aversive agents into transdermal patches to prevent the abuse, diversion, misuse, and accidental exposure of drugs with abuse potential. The AVERSA™ abuse-deterrent technology has the potential to improve the safety profile of transdermal drugs susceptible to abuse, such as fentanyl, while making sure that these drugs remain accessible to those patients who really need them. The technology is covered by a broad intellectual property portfolio with patents granted in the United States, Europe, Japan, Korea, Russia, China, Canada, Mexico, and Australia.
About Nutriband Inc.
We are primarily engaged in the development of a portfolio of transdermal pharmaceutical products. Our lead product under development is an abuse-deterrent fentanyl patch incorporating our AVERSA™ abuse-deterrent technology. AVERSA™ technology can be incorporated into any transdermal patch to prevent the abuse, misuse, diversion, and accidental exposure of drugs with abuse potential.
The Company’s website is www.nutriband.com. Any material contained in or derived from the Company’s websites or any other website is not part of this press release.
About Kindeva
At Kindeva, we manufacture more tomorrows for patients worldwide. With best-in-class facilities and comprehensive CDMO services, we offer more than manufacturing—we deliver strategic value. Our global network of 10 manufacturing and R&D sites offer exceptional integrated knowledge and capabilities, including Annex 1-compliant state-of-the-art aseptic fill finish capacity and next-generation sustainable inhalation propellant technology. By combining expertise in injectable, pulmonary, nasal and dermal drug delivery, we help meet the demands of today and deliver the possibilities of tomorrow. Find out more at https://www.kindevadd.com.
Forward-Looking Statements
Certain statements contained in this press release, including, without limitation, statements containing the words ‘’believes,” “anticipates,” “expects” and words of similar import, constitute “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve both known and unknown risks and uncertainties. The Company’s actual results may differ materially from those anticipated in its forward-looking statements as a result of a number of factors, including those including the Company’s ability to develop its proposed abuse-deterrent fentanyl transdermal system and other proposed products, its ability to obtain patent protection for its abuse technology, its ability to obtain the necessary financing to develop products and conduct the necessary clinical testing, its ability to obtain Federal Food and Drug Administration approval to market any product it may develop in the United States and to obtain any other regulatory approval necessary to market any product in other countries, including countries in Europe, its ability to market any product it may develop, its ability to create, sustain, manage or forecast its growth; its ability to attract and retain key personnel; changes in the Company’s business strategy or development plans; competition; business disruptions; adverse publicity and international, national and local general economic and market conditions and risks generally associated with an undercapitalized developing company, as well as the risks contained under “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in the Company’s Form S-1, Forms 10-K’s and Forms 10-Q’s, and the Company’s other filings with the Securities and Exchange Commission. Except as required by applicable law, we undertake no obligation to revise or update any forward-looking statements to reflect any event or circumstance that may arise after the date hereof.
Patrick McCann, CFA, Research Analyst, Noble Capital Markets, Inc.
Michael Kupinski, Director of Research, Equity Research Analyst, Digital, Media & Technology , Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Hits headwinds in Q2. GoHealth reported Q2 revenue of $94.0 million, below our $110.0 million forecast, as Medicare Advantage softness and CMS policy shifts weighed on volumes. Revenue declined 11% year-over-year. Despite the top-line miss, adj. EBITDA loss of $11.3 million beat our expected loss of $13.2 million, reflecting ongoing cost discipline and benefits from automation initiatives underway in agent workflows.
Recapitalization improves liquidity, alleviates covenant concerns. The company secured $80 million in new term loans and amended its credit agreement to eliminate principal payments through 2026. Liquidity covenants were reduced to a single minimum cash test. While the 4.77 million Class A shares issued represent roughly 20% dilution, we believe the transaction aligns lender and shareholder incentives and resolves the going concern issue.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
NEW YORK, Aug. 07, 2025 (GLOBE NEWSWIRE) — Xcel Brands, Inc. (NASDAQ: XELB), a media and consumer products company known for building influential, creator-led brands, today announced a strategic licensing partnership with TSC Product Lab to launch Mesa Mia by Jenny Martinez—a dynamic new food brand inspired by the bold flavors, rich traditions, and spirit of Latin cooking.
Mesa Mia by Jenny Martinez will offer a curated line of food products designed to bring delicious, authentic meals to the kitchens of Jenny’s fans with ease and flair. The product assortment will include chips, salsas, proteins, meals, drink mixes, and more. This collaboration unites Xcel’s expertise in omnichannel brand development with TSC Product Lab’s strength in product innovation with Jenny’s beloved cooking style.
“We are proud to partner with Jenny and TSC Product Lab to turn her passion and vibrant personality into a brand that will inspire families across the country,” said Robert W. D’Loren, Chairman and CEO of Xcel Brands.
Jenny Martinez—Latina home cook, social media powerhouse, and creator of the brand Mesa Mia by Jenny Martinez—has cultivated a following by sharing colorful, flavorful dishes and heartfelt stories from her kitchen. Her approachable style, combined with a deep love for family and culture, resonates with millions of home cooks seeking connection through food.
“I created Mesa Mia so everyone can bring the joy of Latin cooking into their kitchen with ease. Every Mesa Mia product is a celebration of my family’s traditions and the love we share through food,” said Jenny Martinez. “These are the flavors I grew up with, and I’m so proud to bring a little piece of my cooking into your home.”
Rick Lapine, President of TSC Product Lab, added, “Mesa Mia is a celebration of culture, flavor, and connection—and working with Jenny to bring it to life has been nothing short of inspiring. This is more than a product launch—it’s a movement rooted in love and flavor.”
Mesa Mia Live by Jenny Martinez continues Xcel Brands’ commitment to developing creator-led businesses that speak to how people live, cook, and celebrate today. For more information, please visit www.xcelbrands.com and www.mesamia.com
About Xcel Brands Xcel Brands, Inc. (NASDAQ: XELB) is a media and consumer products company engaged in the design, licensing, marketing, live streaming, and social commerce sales of branded apparel, footwear, accessories, fine jewelry, home goods and other consumer products, and the acquisition of dynamic consumer lifestyle brands. Xcel was founded in 2011 with a vision to reimagine shopping, entertainment, and social media as social commerce. Xcel owns the Halston, Judith Ripka, and C. Wonder brands, as well as the co-branded collaboration brands TowerHill by Christie Brinkley, LB70 by Lloyd Boston, Trust. Respect. Love by Cesar Millan, GemmaMade by Gemma Stafford, and a brand in development with Coco Rocha and also holds noncontrolling interests or long-term license agreements in the Isaac Mizrahi brand, Orme Live and Mesa Mia by Jenny Martinez brands. Xcel also owns and manages the Longaberger brand through its controlling interest in Longaberger Licensing, LLC. Xcel is pioneering a true modern consumer products sales strategy which includes the promotion and sale of products under its brands through interactive television, digital live-stream shopping, social commerce, brick-and-mortar retailers, and e-commerce channels to be everywhere its customers shop. The company’s brands have generated in excess of $5 billion in retail sales via livestreaming in interactive television and digital channels alone and consisting of over 20,000 hours of content production time in live-stream and social commerce. The brand portfolio reaches in excess of 43 million social media followers with broadcast reach into 200 million households. Headquartered in New York City, Xcel Brands is led by an executive team with significant live streaming, production, merchandising, design, marketing, retailing, and licensing experience, and a proven track record of success in elevating branded consumer products companies. For more information, visit www.xcelbrands.com.
AboutTSCLab Products The Sneaky Chef Product Lab (“TSC”) is a cost-effective product development and sourcing company specializing in innovative solutions for the home. The Company’s mission is to create products and brands for leading retailers. Since 2007, TSC has built a diverse portfolio of owned, private label and exclusively licensed brands and has partnered with such legacy names such as Martha Stewart, Sodastream, GreenPan and Calvin Klein. Its network of retail partners includes HSN, QVC, Walmart, Amazon and TJX Companies among others. Led by Rick Lapine, an industry veteran with decades of experience, TSC is supported by a full-time team of passionate experts, bringing over 30 years of combined expertise in sourcing and production. This team has helped establish TSC as a trusted partner for efficient product development, manufacturing, and logistics, with the capability to execute projects rapidly and reliably.
AboutJenny Martinez
Jenny Martinez is well-known across social media platforms for sharing her authentic Mexican family recipes to the delight of millions of fans on TikTok and Instagram. Jenny was born in Mexico and moved to Los Angeles at age four, yet she has never lost touch with her Mexican heritage. The traditional recipes she shares with her followers have been passed down for generations in her family. Although she later mastered her culinary arts on her own, Jenny started by learning most of her mother’s recipes at the age of thirteen. Jenny considers a well-fed family the key to a happy family and believes that dinner should be celebrated every day. Food brings people together, and Jenny’s videos and recipes convey the spirit of family and community. She lives with her family in Los Angeles, California. Jenny’s first cookbook My Mexican Mesa, Y Listo! (Simon & Schuster) debuted in Spring 2024 featuring 100 recipes ranging from breakfast, appetizers & entrees to desserts, and even cocktails! Providing family-style recipes for every occasion & beautifully photographed to capture the authentic spirit of the cuisine, the cookbook is a must-have for home cooks looking for their next delicious meal. In 2024, Jenny launched her own line of cookware and dinnerware/tabletop at over 600 department stores nationally, as well as a spice line sold in grocery stores and online.
Jenny is represented by John Frierson at The Bureau New York City and managed by Lisa Shotland. Her attorney is Phil Daniels at Ginsburg Daniels Kallis LLP
GEO-CM04S1 Offers Solution to Overcome HHS-Cited mRNA Deficiencies – Potentially Offering Broader, More Durable Protection
ATLANTA, GA – August 7, 2025 – GeoVax Labs, Inc. (Nasdaq: GOVX), a clinical-stage biotechnology company developing multi-antigen, MVA-based vaccines and solid tumor immunotherapies, today issued a statement in response to the U.S. Department of Health and Human Services’ (HHS) decision to terminate nearly $500 million in BARDA-funded mRNA vaccine development contracts.
This action reflects a policy shift, underscored by HHS Secretary Kennedy addressing fundamental concerns around mRNA vaccines. In a post on X, Secretary Kennedy stated: “mRNA vaccines don’t perform well against viruses that infect the upper respiratory tract”. The Secretary added that this is due to a concept known as “antigenic shift, meaning that the vaccine paradoxically encourages new mutations and can actually prolong pandemics as the virus constantly mutates”. GeoVax’s vaccine candidates, including GEO-CM04S1 for COVID-19, are designed to induce immunity using multiple antigens. GEO-CM04S1 expresses both the Spike (S) and Nucleocapsid (N) proteins of SARS-CoV-2, enabling broader and more durable protection – even as the virus mutates. Data from clinical studies have demonstrated that GEO-CM04S1 induces immune responses across variants, from the original Wuhan strain through Omicron, even in immunocompromised patients.
“Secretary Kennedy’s remarks spotlight the exact issue our platform was designed to overcome,” said David Dodd, Chairman and CEO of GeoVax. “The mRNA approach, with its single-target design, appears limited relative to durability and antigenic shift. GEO-CM04S1’s multi-antigen construct represents a promising solution – with clinical data demonstrating broader immunity and addressing the mRNA shortcomings Kennedy described, including among the most vulnerable immunocompromised patients.”
Why GeoVax’s MVA-Based Vaccines Align with U.S. Priorities
Multi‑antigen breadth and durability – GeoVax’s COVID-19 vaccine candidate, GEO‑CM04S1, expresses both SARS‑CoV‑2 Spike and Nucleocapsid proteins – inducing robust and durable antibody and T‑cell immunity in Phase 2 trials. Notably, in a trial among Chronic Lymphocytic Leukemia (CLL) patients, the mRNA comparator arm was halted for failing to meet immune‑response benchmarks, while GEO‑CM04S1 exceeded interim endpoints, with the remainder of the ongoing study only including the GEO-CM04S1 arm.
Safety for vulnerable populations – MVA does not replicate in human cells and has been FDA‑approved for use in immunocompromised individuals, pregnant women, and children. This makes it a validated vaccine platform with broad safety.
Manufacturing innovation and U.S. resilience – GeoVax is advancing AGE.1 manufacturing processes for MVA-based vaccines such as GEO-CM04S1 which is expected to support scalable, decentralized U.S. vaccine manufacturing, providing faster production, higher yield at a reduced cost – a strategic advantage for public health resilience.
Broad infectious-disease pipeline – Beyond COVID‑19, GeoVax is pursuing MVA‑based vaccines targeting hemorrhagic fever viruses (Ebola Zaire, Ebola Sudan, Marburg), Zika, and Mpox/Smallpox. This diversified pipeline aligns with HHS, NIH, FDA, and WHO priorities for pandemic preparedness and biodefense.
GeoVax: Delivering on the “Safer, Broader” Vision
GeoVax applauds the call to shift toward multi-antigen vaccine designs with validated safety profiles and resilience to viral mutation. The MVA‑based platform delivers on these goals:
Evidence‑backed safety for immunocompromised, pediatric, and pregnant populations.
Broader immune responses with Spike plus Nucleocapsid antigens.
Strategic manufacturing design for domestic scale and surge capacity.
Multi‑disease readiness across COVID‑19, hemorrhagic fevers, Zika, and Mpox/Smallpox.
Next Steps: Turning Rhetoric into Resilience
GeoVax urges HHS to proactively support robust, evidence-backed alternatives – including multi-antigen platforms like MVA as part of a diversified, resilient biomedical countermeasure arsenal.
GeoVax invites HHS and other federal partners to collaborate under the FDA’s Commissioner’s National Priority Voucher (CNPV) program and other mechanisms to accelerate regulatory and funding support for GEO-CM04S1 and GEO-MVA, both of which are uniquely positioned to protect high-risk populations and enhance U.S. biomanufacturing self-sufficiency.
Dodd added: “We must now invest in platforms that reflect what we’ve learned. GeoVax stands ready to lead this transition. Our MVA platform already has a safety track record and is delivering early efficacy where mRNA appears to have fallen short. We stand ready and prepared to assist in supporting HHS’s bold reset into a long-term strategy for national immunization security.”
About GeoVax
GeoVax Labs, Inc. is a clinical-stage biotechnology company developing novel vaccines against infectious diseases and therapies for solid tumor cancers. The Company’s lead clinical program is GEO-CM04S1, a next-generation COVID-19 vaccine currently in three Phase 2 clinical trials, being evaluated as (1) a primary vaccine for immunocompromised patients such as those suffering from hematologic cancers and other patient populations for whom the current authorized COVID-19 vaccines are insufficient, (2) a booster vaccine in patients with chronic lymphocytic leukemia (CLL) and (3) a more robust, durable COVID-19 booster among healthy patients who previously received the mRNA vaccines. In oncology the lead clinical program is evaluating a novel oncolytic solid tumor gene-directed therapy, Gedeptin®, having recently completed a multicenter Phase 1/2 clinical trial for advanced head and neck cancers. GeoVax is also developing a vaccine targeting Mpox and smallpox and, based on recent EMA regulatory guidance, anticipates progressing directly to a Phase 3 clinical evaluation, omitting Phase 1 and Phase 2 trials. GeoVax has a strong IP portfolio in support of its technologies and product candidates, holding worldwide rights for its technologies and products. For more information about the current status of our clinical trials and other updates, visit our website: www.geovax.com.
Forward-Looking Statements
This release contains forward-looking statements regarding GeoVax’s business plans. The words “believe,” “look forward to,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “could,” “target,” “potential,” “is likely,” “will,” “expect” and similar expressions, as they relate to us, are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. Actual results may differ materially from those included in these statements due to a variety of factors, including whether: GeoVax is able to obtain acceptable results from ongoing or future clinical trials of its investigational products, GeoVax’s immuno-oncology products and preventative vaccines can provoke the desired responses, and those products or vaccines can be used effectively, GeoVax’s viral vector technology adequately amplifies immune responses to cancer antigens, GeoVax can develop and manufacture its immuno-oncology products and preventative vaccines with the desired characteristics in a timely manner, GeoVax’s immuno-oncology products and preventative vaccines will be safe for human use, GeoVax’s vaccines will effectively prevent targeted infections in humans, GeoVax’s immuno-oncology products and preventative vaccines will receive regulatory approvals necessary to be licensed and marketed, GeoVax raises required capital to complete development, there is development of competitive products that may be more effective or easier to use than GeoVax’s products, GeoVax will be able to enter into favorable manufacturing and distribution agreements, and other factors, over which GeoVax has no control.
Further information on our risk factors is contained in our periodic reports on Form 10-Q and Form 10-K that we have filed and will file with the SEC. Any forward-looking statement made by us herein speaks only as of the date on which it is made. Factors or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of them. We undertake no obligation to publicly update any forward-looking statement, whether as a result of new information, future developments or otherwise, except as may be required by law.
CHICAGO, Aug. 07, 2025 (GLOBE NEWSWIRE) — GoHealth, Inc. (NASDAQ: GOCO) (“GoHealth” or the “Company”), a leading health insurance marketplace and Medicare-focused digital health company, today announced the execution of strategic capital and governance actions that are expected to significantly enhance its financial flexibility and long-term positioning along with its financial results for the three and six months ended June 30, 2025.
Strategic Capital and Governance Actions
Secured new senior secured superpriority term loan facility, including $80.0 million in new-money term loans and $35.0 million in roll-up loans, to support working capital and strategic flexibility heading into the Medicare annual enrollment period.
Expect additional liquidity in the term loan to allow us to maintain compliance with our debt covenants and fund our operations for the next 12 months and beyond.
Amended existing credit agreement to waive near-term principal payments through 2026 and reset financial covenants.
Created debt basket capacity of up to $250.0 million under the new superpriority term loan facility and amended credit agreement, to pursue potential transformative transactions.
Issued an aggregate of 4,766,219 shares of Class A common stock to lenders, reinforcing alignment with long-term stockholder value creation.
Appointed three new directors to the Company’s board of directors and accepted resignations from three existing directors to align governance with GoHealth’s forward-looking strategic direction.
“Our strategic capital and governance actions reflect our commitment to long-term stockholder value creation and our belief that GoHealth is structurally and strategically positioned to lead in a consolidating industry,” said Vijay Kotte, CEO of GoHealth. “With the new credit facility and the access to immediate and expandable capital it provides, we believe we are operating from a position of strength as we continue to serve the Medicare market, pursue disciplined growth and assess transformative opportunities.”
“The amendment to our existing credit agreement provides important financial flexibility,” said Brendan Shanahan, CFO of GoHealth. “Through this strategic financing arrangement, we have the ability to evaluate and pursue strategic transactions. We believe these enhancements position us to act decisively and responsibly in support of our strategic objectives.”
Additional information, including with respect to the Company’s preliminary financial results for the three and six months ended June 30, 2025, is included in the tables at the end of this press release.
Conference Call Details
The Company will host a conference call today, Thursday, August 7, 2025 at 8:00 a.m. (ET) to discuss recent strategic capital and governance actions. A live audio webcast of the conference call will be available via GoHealth’s Investor Relations website, https://investors.gohealth.com/. A replay of the call will be available via webcast for on-demand listening shortly after the completion of the call.
About GoHealth, Inc.
GoHealth is a leading health insurance marketplace and Medicare-focused digital health company whose purpose is to compassionately ensure consumers’ peace of mind when making healthcare decisions so they can focus on living life. For many of these consumers, enrolling in a health insurance plan is confusing and difficult, and seemingly small differences between health plans may lead to significant out-of-pocket costs or lack of access to critical providers and medicines. GoHealth’s proprietary technology platform leverages modern machine-learning algorithms, powered by over two decades of insurance purchasing behavior, to reimagine the process of matching a health plan to a consumer’s specific needs. Its unbiased, technology-driven marketplace coupled with highly skilled licensed agents has facilitated the enrollment of millions of consumers in Medicare plans since GoHealth’s inception. For more information, visit https://www.gohealth.com.
Investor Relations:
John Shave
JShave@gohealth.com
Media Relations:
Pressinquiries@gohealth.com
Forward-Looking Statements
This press release contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These forward-looking statements are made in reliance upon the safe harbor provision of the Private Securities Litigation Reform Act of 1995. All statements other than statements of historical facts contained in this press release may be forward-looking statements. Statements regarding our future results of operations and financial position, liquidity, business strategy and plans and objectives of management for future operations, including, among others, statements regarding the expected results of the strategic capital and governance actions announced hereby, our expected growth, pursuit of strategic alternatives and strategic objectives, long-term value creation, magnitude of expected impairments, our capital structure, future capital expenditures, debt service obligations, ability to continue as a going concern, adoption and use of artificial intelligence technologies, the impact on our business from regulatory changes, the impact on our business from the acquisition of e-TeleQuote Insurance, Inc. (“e-TeleQuote”) and our ability to successfully integrate e-TeleQuote’s operations, technologies and employees into our business, are forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “aims,” “expects,” “plans,” “anticipates,” “could,” “intends,” “targets,” “projects,” “contemplates,” “believes,” “estimates,” “predicts,” “potential,” “likely,” “future” or “continue” or the negative of these terms or other similar expressions. The forward-looking statements in this press release are only predictions, projections and other statements about future events that are based on current expectations and assumptions. Accordingly, we caution you that any such forward-looking statements are not guarantees of future performance and are subject to risks, assumptions and uncertainties that are difficult to predict. Although we believe that the expectations reflected in these forward-looking statements are reasonable as of the date made, actual results may prove to be materially different from the results expressed or implied by the forward-looking statements.
These forward-looking statements speak only as of the date of this press release and are subject to a number of important factors that could cause actual results to differ materially from those in the forward-looking statements, including our inability to realize our expectations with respect to the strategic capital and governance actions announced hereby, our inability to execute on strategic alternatives or strategic objectives, our level of indebtedness, our level of liquidity, and the factors described in the sections titled “Summary Risk Factors,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Annual Report on Form 10-K for the fiscal year ended December 31, 2024 (“2024 Annual Report on Form 10-K”), our Quarterly Report on Form 10-Q for the first quarter ended March 31, 2025 (“Q1 2025 Quarterly Report on Form 10-Q”), our forthcoming Quarterly Report on Form 10-Q for the second quarter ended June 30, 2025 (“Q2 2025 Quarterly Report on Form 10-Q”) and in our other filings with the Securities and Exchange Commission. The factors described in our 2024 Annual Report on Form 10-K, our Q1 2025 Quarterly Report on Form 10-Q and our forthcoming Q2 2025 Quarterly Report on Form 10-Q should not be construed as exhaustive and should be read together with the other cautionary statements included in this press release, as well as the cautionary statements and other risk factors set forth in our other filings with the Securities and Exchange Commission.
You should read this press release and the documents that we reference in this press release completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
DLH delivers improved health and readiness solutions for federal programs through research, development, and innovative care processes. The Company’s experts in public health, performance evaluation, and health operations solve the complex problems faced by civilian and military customers alike, leveraging digital transformation, artificial intelligence, advanced analytics, cloud-based applications, telehealth systems, and more. With over 2,300 employees dedicated to the idea that “Your Mission is Our Passion,” DLH brings a unique combination of government sector experience, proven methodology, and unwavering commitment to public health to improve the lives of millions. For more information, visit www.DLHcorp.com.
Joe Gomes, CFA, Managing Director, Equity Research Analyst, Generalist , Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Making Progress. In the third quarter, DLH effectively navigated changes in the competitive landscape and transition in the industry overall, preserving margin delivery and strong operating cash flow. Headwinds such as the transition of CMOP locations, unbundling of DOD contracts, and scope reductions as a result of government efficiency efforts all impacted the quarter.
3Q25 Results. Revenue was $83.3 million, compared to $100.7 million in the year ago quarter. We had forecasted $83 million. DLH reported adjusted EBITDA of $8.1 million, down from $10 million in 3Q24 and our $8.5 million estimate. Net income was $0.3 million, or $0.02/sh, versus $1.1 million, or $0.08/sh last year. We had projected $0.35 million, or $0.02/sh.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Phase 1b Kidney Transplant Data Presented. Eledon presented data from its Phase 1b trial using tegoprubart as part of an immunosuppressive regimen at The World Transplant Congress. The data from the first 32 patients at two dosage cohorts continues to show meaningful improvement over the standard of care. We believe this supports our expectations for strong data for the Phase 2 BESTOW trial in November.
Study Design. The presentation included data from 32 patients receiving kidney transplants followed by an immunosuppressive regimen tegoprubart instead of tacrolimus, the standard of care. The primary endpoints are safety and pharmacokinetics. Secondary endpoints include patient survival, graft survival, biopsy proven acute rejection, with kidney function measured by eGFR and iBOX score.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Robert LeBoyer, Senior Vice President, Equity Research Analyst, Biotechnology, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Cadrenal Announces New Trial Design. Cadrenal announced that it plans to begin a trial testing tecarfarin in patients who are starting renal dialysis, both with and without atrial fibrillation (ESKD-Afib). This design reflects recent studies showing that the first several months after starting dialysis are an ultra-high risk period for mortality and cardiac events. The trial will test tecarfarin efficacy in reducing these events and could begin in late 2025 to early 2026.
Modified Study Design Focuses On Highest Risk Period. The initiation of renal dialysis impacts several important cardiovascular and renal functions. New studies show that the first six months after starting dialysis have a 20-fold increase in cardiovascular events and mortality. This has not previously been recognized due to pathologies of the underlying conditions that lead to CKD and dialysis.
Equity Research is available at no cost to Registered users of Channelchek. Not a Member? Click ‘Join’ to join the Channelchek Community. There is no cost to register, and we never collect credit card information.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
NORWOOD, Mass., Aug. 06, 2025 (GLOBE NEWSWIRE) — MariMed Inc. (“MariMed” or the “Company”) (CSE: MRMD) (OTCQX: MRMD), a leading multi-state cannabis operator focused on improving lives every day, today announced its financial results for the second quarter ended June 30, 2025.
Management Commentary
“We delivered growth and expanded operations across our business during the second quarter, continuing our progress of building a leading cannabis consumer packaged goods company,” said Jon Levine, MariMed Chief Executive Officer. “Our ‘Expand the Brand’ strategy is working. Our innovative, high-quality portfolio of brands grew or maintained their market share across our core markets. We remain confident in delivering the shareholder value our investors deserve by leveraging our brands as the primary growth engine of our company. Looking ahead, we anticipate increasing product distribution through the addition of adult-use sales in Delaware, a new licensing agreement in Maine, and our recently announced entry into Pennsylvania. In addition, the strength of our balance sheet affords us optionality with respect to M&A and licensing opportunities.”
“We delivered sequential growth in both wholesale and retail revenues for the second quarter, a substantial increase in adjusted EBITDA, and we were cash flow positive,” said Mario Pinho, MariMed Chief Financial Officer. “Our performance reflects strong execution in Massachusetts, full-quarter contributions from Delaware, and a solid retail strategy. With the METRC system migration in Illinois behind us and Missouri under active review, we remain confident in the revenue catalysts we have built for the second half of the year, including adult use in Delaware, entry into Pennsylvania, and expanded wholesale.”
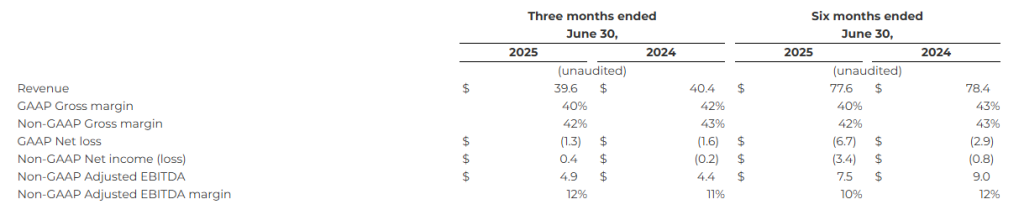
Financial Highlights1
The following table summarizes the Company’s consolidated financial highlights (in millions, except percentage amounts):
1 See the reconciliations of non-GAAP financial measures to the most directly comparable GAAP measures and additional information about non-GAAP measures in the section entitled “Discussion of Non-GAAP Financial Measures” below and in the financials information included herewith.
CONFERENCE CALL
MariMed management will host a conference call on Thursday, August 7, 2025 at 8:00 a.m. Eastern time, to discuss these results. The conference call may be accessed through MariMed’s Investor Relations website, or by clicking the following link: Q2 2025 MRMD Earnings Call.
SECOND QUARTER 2025 OPERATIONAL HIGHLIGHTS
During the second quarter, the Company announced the following development in the implementation of its strategic growth plan:
April 1: Launched its Nature’s Heritage™-branded cannabis flower, pre-rolls, and vapes in Illinois, marking the first time the brand’s premium products are available in the state.
April 3: Expanded the line-up of its top-selling Betty’s Eddies™-branded cannabis chews with the introduction of a new caramel chew, Betty’s Caramelt Away.
April 8: Promoted Ryan Crandall to Chief Commercial Officer to lead the Company’s commercial strategy and activities, including Sales, Marketing, Product Development, and Retail Operations. He had served as the Company’s Chief Revenue Officer since July 2022, and previously was its Chief Products Officer and SVP, Sales for four years.
May 29: Expended its branded product line-up with the introduction of MycroDose by Nature’s Heritage, a vegan pill that combines full-spectrum cannabis with the added benefits of functional mushrooms.
OTHER DEVELOPMENTS
Subsequent to the end of the second quarter, the Company announced the following further developments:
July 14: Expanded the distribution of Betty’s Eddies to Maine for both adult-use cannabis consumers and medical patients through a new licensing partnership.
July 31: Announced a Managed Services Agreement (“MSA”) to assume day-to-day management of a cultivation and processing facility in Pennsylvania owned by a division of multi-state cannabis operator TILT Holdings. In addition, a licensing agreement will enable the Company to distribute its award-winning, branded products in Pennsylvania, which is anticipated to become the next state to expand its legal cannabis program to include adult-use sales.
DISCUSSION OF NON-GAAP FINANCIAL MEASURES
MariMed’s management uses several different financial measures, both GAAP and non-GAAP, in analyzing and assessing the overall performance of its business, making operating decisions, and planning and forecasting future periods. The Company has provided in this release several non-GAAP financial measures: Non-GAAP Adjusted EBITDA and non-GAAP Adjusted EBITDA margin, Non-GAAP Gross margin, Non-GAAP Operating expenses and Non-GAAP Net income (loss), as supplements to Revenue, Gross margin, Operating expenses, Income (loss) from operations, Net income (loss) and other financial measures prepared in accordance with GAAP.
Management believes these non-GAAP financial measures are useful in reviewing and assessing the performance of the Company, and when planning and forecasting future periods, as they provide meaningful operating results by excluding the effects of expenses that are not reflective of its operating business performance. In addition, the Company’s management uses these non-GAAP financial measures to understand and compare operating results across accounting periods and for financial and operational decision-making. The presentation of these non-GAAP measures is not intended to be considered in isolation or as a substitute for the financial information prepared in accordance with GAAP.
Management believes that investors and analysts benefit from considering non-GAAP financial measures in assessing the Company’s financial results and its ongoing business, as it allows for meaningful comparisons and analysis of trends in the business. In particular, non-GAAP adjusted EBITDA is used by many investors and analysts themselves, along with other metrics, to compare financial results across accounting periods and to those of peer companies.
As there are no standardized methods of calculating non-GAAP financial measures, the Company’s calculations may differ from those used by analysts, investors and other companies, even those within the cannabis industry, and therefore may not be directly comparable to similarly titled measures used by others.
Management defines non-GAAP Adjusted EBITDA as income (loss) from operations, determined in accordance with GAAP, excluding the following items:
depreciation and amortization of property and equipment;
amortization of acquired intangible assets;
impairment or write-downs of acquired intangible assets;
inventory revaluation;
stock-based compensation;
severance;
legal settlements; and
acquisition-related and other expenses.
For further information, please refer to the publicly available financial filings available on MariMed’s Investor Relations website, as filed with the U.S. Securities and Exchange Commission, or as filed with the Canadian securities regulatory authorities on the SEDAR website.
ABOUT MARIMED MariMed Inc. is a leading multi-state cannabis operator, known for developing and managing state-of-the-art cultivation, production, and retail facilities. Our award-winning portfolio of cannabis brands, including Betty’s Eddies™, Bubby’s Baked™, Vibations™, InHouse™, and Nature’s Heritage™, sets us apart as an industry leader. These trusted brands, crafted with quality and innovation, are recognized and loved by consumers across the country. With a commitment to excellence, MariMed continues to drive growth and set new standards in the cannabis industry. For additional information, visit www.marimedinc.com.
IMPORTANT CAUTION REGARDING FORWARD-LOOKING STATEMENTS: The information in this release contains “forward-looking” statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995, which are subject to several risks and uncertainties. All statements other than statements of historical facts contained in this release, including without limitation statements regarding projected financial results for 2025, including anticipated openings of dispensaries and facilities, timing of regulatory approvals, plans and objectives of management for future operations, are forward-looking statements. Without limiting the foregoing, the words “anticipates”, “believes”, “estimates”, “expects”, “expectations”, “intends”, “may”, “plans”, and other similar language, whether in the negative or affirmative, are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words.
Forward-looking statements are based on our current beliefs and assumptions regarding our business, timing of regulatory approvals, the ability to obtain new licenses, business prospects and strategic growth plan, and other future conditions. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that are difficult to predict. Our actual results may differ materially from those contemplated in these forward-looking statements due to various risks, uncertainties, and other important factors, including, among others, reductions in customer spending, our ability to recruit and retain key personnel, and disruptions from the integration efforts of acquired companies.
These factors are not intended to be an all-encompassing list of risks and uncertainties that may affect our business and results of operations. These statements are not a guarantee of future performance and involve risk and uncertainties that are difficult to predict, including, among other factors, changes in demand for the Company’s services and products, changes in the law and its enforcement, and changes in the economic environment. Additional information regarding these and other factors can be found in our reports filed with the U.S. Securities and Exchange Commission. In providing these forward-looking statements, the Company expressly disclaims any obligation to update these statements publicly or otherwise, whether as a result of new information, future events or otherwise, except as required by law.
All trademarks and service marks are the property of their respective owners.
Neither the CSE nor its Regulation Services accepts responsibility for the adequacy or accuracy of this release.
For More Information Contact:
Howard Schacter, Chief Communications Officer Email: hschacter@marimedinc.com Phone: (781) 277-0007
Data from patients who remained on tegoprubart for a year showed overall mean 12-month eGFR of approximately 68 mL/min/1.73 m² post-transplant
Preliminary iBox data, a key biomarker of kidney function and immunologic response, supports that tegoprubart may improve 5-year graft survival vs. current standard of care
Tegoprubart continues to be well tolerated with no cases of death, graft loss, drug related tremor, or new-onset diabetes
Conference call to be held today at 4:30 p.m. ET
IRVINE, Calif., Aug. 06, 2025 (GLOBE NEWSWIRE) — Eledon Pharmaceuticals, Inc. (“Eledon”) (NASDAQ: ELDN) today announced updated data from the Company’s ongoing open-label Phase 1b trial evaluating tegoprubart for the prevention of organ rejection in kidney transplant patients. Results from the oral presentation, titled “Tegoprubart, an Anti-CD40L Antibody, for the Prevention of Rejection in Kidney Transplantation: An Ongoing Phase 1b Study,” were presented today at the World Transplant Congress (WTC) taking place in San Francisco, CA.
“The data presented today at WTC further reinforce our belief that tegoprubart has the potential to not only provide better protection and long-term preservation of kidney function following transplantation, but also to offer a safer alternative to traditional immunosuppressive therapies by minimizing harmful side effects,” said David-Alexandre C. Gros, M.D., Chief Executive Officer of Eledon. “The continued strength of the Phase 1b data through 12 months of treatment is highly encouraging as we look ahead to topline results from our Phase 2 BESTOW trial, expected in November, which compares tegoprubart to tacrolimus, the current standard of care.”
As of the July 2025 cutoff date, 32 patients undergoing kidney transplantation have been enrolled in the Phase 1b study. Updated data showed that kidney function, as assessed by estimated glomerular filtration rate (eGFR), stabilized after the first month post-transplant and remained in the range of approximately 68 mL/min/1.73 m2 through 12 months for patients (n=12) who remained on tegoprubart. Kidney function in the intention-to-treat population (n=15) was approximately 63 mL/min/1.73 m2 at 12 months. Data from historical studies using the standard of care, calcineurin inhibitor-based immunosuppression therapy, typically report aggregate mean estimated glomerular filtration rates (eGFRs) of approximately 53 mL/min/1.73 m2 during the first year after kidney transplant.
In addition, preliminary abbreviated iBox data was presented suggesting that tegoprubart may improve 5-year graft survival. Abbreviated iBox, a composite biomarker panel developed by the Paris Transplant Group, incorporates kidney function (eGFR, proteinuria) and immunologic response (donor-specific antibodies) parameters into a single prognostic score. Based on data collected to date, abbreviated iBox scores were -3.75 in the intention-to-treat population and -4.11 in the on-treatment population, which compare favorably to a -2.98 historical mean for calcineurin inhibitors. A difference in abbreviated iBox score of -0.40 at 12 months is considered predictive of a 4-5% difference in 5-year graft survival suggesting that tegoprubart may have a predicted 5-year allograft survival rate of over 96%.
Mean tegoprubart treatment exposure to date was 233 days. Tegoprubart continues to be well-tolerated with no cases of death, graft loss, drug related tremor, or new-onset diabetes, a side effect associated with standard of care immunosuppression therapy.
There were six (18.8%) rejection episodes, and 75% of patients who experienced a rejection had received low-dose rabbit antithymocyte globulin (rATG) induction. All rejection episodes were successfully treated. Of the patients who experienced a rejection episode and completed a year in the study, three who remained on tegoprubart had a mean eGFR of approximately 73 mL/min/1.73 m2 at 12 months, indicating full recovery of kidney function, while the two patients who switched to standard of care tacrolimus had a mean eGFR of approximately 34 mL/min/1.73 m² at 12 months.
All 32 patients received rATG induction therapy and a maintenance regimen consisting of tegoprubart, mycophenolate mofetil, and corticosteroids.
Cohort 1 has completed enrollment and evaluated tegoprubart at a dose of 20 mg/kg with rATG induction up to 6 mg/kg.
Cohort 2 is currently enrolling and is evaluating a lower tegoprubart dose of 10 mg/kg, with a required rATG dose of 4.5 mg/kg.
The primary endpoint of the study is safety and pharmacokinetics. Secondary and exploratory endpoints include patient and graft survival, biopsy-proven acute rejection, kidney function as measured by estimated by eGFR, and abbreviated iBox score.
Eledon is also conducting a Phase 2 trial (BESTOW; NCT05983770) and a long-term safety and efficacy extension study (NCT06126380) to evaluate tegoprubart for the prevention of organ rejection in patients receiving a kidney transplant. Topline results from the Phase 2 BESTOW trial are anticipated in November 2025.
Full details of the WTC oral presentation are below:
Title: Tegoprubart, an Anti-CD40L Antibody, for the Prevention of Rejection in Kidney Transplantation: An Ongoing Phase 1b Study Session: Oral Presentation, Kidney Novel Immunosuppressant Strategies Presenter: John Gill, MD, MS, University of British Columbia, Vancouver, Canada Session Date and Time: Wednesday, August 6, 2025: 10:00 a.m. – 11:15 a.m. PT
Conference Call
Eledon will hold a conference call today, August 6, 2025 at 4:30 p.m. Eastern Time to discuss the updated Phase 1b trial results. To join the conference call, please dial 1-800-717-1738 for domestic callers or 1-646-307-1865 for international callers. The conference ID is 34575. Registration for the live webcast can be found here and available on the “Events” section of Eledon’s website at www.eledon.com. The webcast will be archived on the website following the completion of the call.
About Eledon Pharmaceuticals and tegoprubart
Eledon Pharmaceuticals, Inc. is a clinical stage biotechnology company that is developing immune-modulating therapies for the management and treatment of life-threatening conditions. The Company’s lead investigational product is tegoprubart, an anti-CD40L antibody with high affinity for the CD40 Ligand, a well-validated biological target that has broad therapeutic potential. The central role of CD40L signaling in both adaptive and innate immune cell activation and function positions it as an attractive target for non-lymphocyte depleting, immunomodulatory therapeutic intervention. The Company is building upon a deep historical knowledge of anti-CD40 Ligand biology to conduct preclinical and clinical studies in kidney allograft transplantation, xenotransplantation, and amyotrophic lateral sclerosis (ALS). Eledon is headquartered in Irvine, California. For more information, please visit the Company’s website at www.eledon.com.
Follow Eledon Pharmaceuticals on social media: LinkedIn; Twitter
About iBox
iBox is a composite biomarker panel developed by the Paris Transplant Group to predict long-term kidney graft survival. It combines kidney function (eGFR, proteinuria), immunologic response (donor-specific antibodies), and histopathology (Banff scores) into a single prognostic score. Validated across four independent cohorts, including two Phase 3 trials (BMS BENEFIT and BENEFIT-EXT), iBox has demonstrated strong predictive accuracy (C-statistic >0.8) for 5-year graft loss and outperforms traditional markers like biopsy-proven acute rejection. Both full and abbreviated iBox models have been qualified by the European Medicines Agency (EMA) and accepted by the U.S. FDA into the Biomarker Qualification Program. The iBox Composite Biomarker Panel is under review by the FDA as a Reasonably Likely Surrogate Endpoint (RLSE) for use as a co-primary endpoint in Phase 2/3 trials, supporting potential accelerated approval of novel immunosuppressive therapies. This makes iBox the first transplant-specific endpoint formally recognized under FDA’s biomarker qualification framework.
Forward-Looking Statements
This press release contains forward-looking statements that involve substantial risks and uncertainties. Any statements about the company’s future expectations, plans and prospects, including statements about ongoing clinical trials, the development of product candidates, expected timing for initiation of future clinical trials, expected timing for receipt of data from clinical trials, expected or future results of tegoprubart trials and its ability to prevent rejection in connection with kidney transplantation, as well as other statements containing the words “believes,” “anticipates,” “plans,” “expects,” “estimates,” “intends,” “predicts,” “projects,” “targets,” “looks forward,” “could,” “may,” and similar expressions, constitute forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements are inherently uncertain and are subject to numerous risks and uncertainties, including: risks relating to the safety and efficacy of our drug candidates; risks relating to clinical development timelines, including interactions with regulators and clinical sites, as well as patient enrollment; and risks relating to costs of clinical trials and the sufficiency of the company’s capital resources to fund planned clinical trials. Actual results may differ materially from those indicated by such forward-looking statements as a result of various factors. These risks and uncertainties, as well as other risks and uncertainties that could cause the company’s actual results to differ significantly from the forward-looking statements contained herein, are discussed in our quarterly 10-Q, annual 10-K, and other filings with the U.S. Securities and Exchange Commission, which can be found at www.sec.gov. Any forward-looking statements contained in this press release speak only as of the date hereof and not of any future date, and the company expressly disclaims any intent to update any forward-looking statements, whether as a result of new information, future events or otherwise.
Follow Eledon Pharmaceuticals on social media: LinkedIn; Twitter
Alcon (NYSE: ALC), a global leader in eye care, has signed a definitive agreement to acquire STAAR Surgical Company (NASDAQ: STAA) in a cash transaction valued at approximately $1.5 billion. The acquisition is aimed at bolstering Alcon’s position in the surgical vision correction market, particularly in addressing the growing global demand for alternatives to LASIK.
The deal will see Alcon purchasing all outstanding shares of STAAR common stock at $28 per share, representing a 59% premium to STAAR’s 90-day volume-weighted average price and a 51% premium over its August 4 closing price.
STAAR Surgical is best known for its EVO family of Implantable Collamer Lenses (ICLs), which offer minimally invasive, reversible vision correction for patients with moderate to high myopia, including those with astigmatism. These lenses are implanted behind the iris and in front of the eye’s natural lens, offering a surgical option that avoids corneal tissue removal.
For Alcon, the acquisition is a strategic complement to its existing laser vision correction business. By incorporating STAAR’s EVO ICL technology, the company aims to provide a broader spectrum of refractive solutions for patients, especially those who are not ideal candidates for LASIK or other laser procedures.
The need for such alternatives is expanding rapidly. Global studies suggest that by 2050, half of the world’s population will be myopic, with approximately 500 million people falling into the high myopia category—a group that often requires advanced vision correction techniques.
Alcon expects the acquisition to be accretive to earnings by the second year post-closing. The company plans to finance the purchase through short- and long-term credit facilities and noted that the transaction is not subject to a financing condition.
STAAR has faced recent market challenges, including fluctuating demand in key international markets such as China. By joining Alcon, STAAR is expected to benefit from increased operational scale and broader global distribution, which could accelerate the adoption of its EVO ICLs.
The transaction has received unanimous approval from both companies’ boards of directors. It is expected to close within six to twelve months, pending customary closing conditions, including regulatory clearances and approval by STAAR shareholders.
Financial advisors on the deal include Morgan Stanley for Alcon and Citi for STAAR, while legal counsel was provided by Gibson, Dunn & Crutcher LLP and Wachtell, Lipton, Rosen & Katz, respectively.
As part of ongoing developments, STAAR is scheduled to release its Q2 2025 earnings on August 6, though it will not hold an investor conference call due to the pending acquisition.