EL SEGUNDO, Calif.–(BUSINESS WIRE)– The Beachbody Company, Inc. (NASDAQ: BODI) (“BODi” or the “Company”), a leading fitness and nutrition company, will release its fourth quarter 2025 results on Tuesday, March 10, 2026, after the U.S. stock market closes. The Company will host a conference call at 5:00 p.m. (Eastern Time) that day to discuss the results.
For those unable to participate in the conference call, a replay will be available after the conclusion of the call on March 10, 2026, through March 17, 2026. The toll-free replay dial-in number is (866) 813-9403 (U.S & Canada). The replay passcode is 989620.
About BODi and The Beachbody Company, Inc.
BODi, formerly known as Beachbody, has been a pioneer in structured, step-by-step home fitness and nutrition programs for nearly three decades, with iconic products such as P90X, INSANITY, 21 Day Fix and the original premium superfood nutrition supplement, Shakeology. Since its inception, BODi has helped more than 30 million people reach life-changing results. Today, BODi continues to evolve with a simple mission: help people achieve their goals and lead healthier, more fulfilling lives, especially busy, time-strapped people who want to fit healthy habits into everyday life with proven solutions. The BODi community empowers millions to stay motivated and accountable, supporting healthy weight management, improved metabolic function, increased mental and physical well-being, better sleep, as well as evidence-based habits that enhance healthspan and longevity.
NEW ALBANY, Ohio, Feb. 24, 2026 (GLOBE NEWSWIRE) — Commercial Vehicle Group (the “Company” or “CVG”) (NASDAQ: CVGI) will hold its quarterly conference call on Wednesday, March 11, 2026, at 8:30 a.m. ET, to discuss fourth quarter and full year 2025 financial results. CVG will issue a press release and presentation prior to the conference call.
Toll-free participants dial (800) 549-8228 using conference code 51917. International participants dial (289) 819-1520 using conference code 51917. This call is being webcast and can be accessed through the “Investors” section of CVG’s website at ir.cvgrp.com where it will be archived for one year.
A telephonic replay of the conference call will be available until March 25, 2026. To access the replay, toll-free callers can dial (+1) 888 660 6264 using access code 51917 #, and toll callers in North America and other locations can dial (+1) 289 819 1325.
About CVG
At CVG, we deliver real solutions to complex design, engineering and manufacturing problems while creating positive change for our customers, industries, and communities we serve. Information about the Company and its products is available on the internet at www.cvgrp.com.
Investor Relations Contact: Ross Collins or Nathan Skown Alpha IR Group CVGI@alpha-ir.com
Reported positive update from Phase 2 CRDF-004 trial in first-line RAS-mutated mCRC, with the 30 mg onvansertib + FOLFIRI/bev arm demonstrating: • Robust ORR of 72.2% (vs 43.2% with combined SoC of FOLFOX/bev and FOLFIRI/bev) • Significant improvement in PFS over combined SoC (HR: 0.37, p<0.05)
Data support selection of 30 mg onvansertib dose in combination with FOLFIRI/bev for planned registrational program; detailed data and registrational plans expected in the first half of 2026
SAN DIEGO, Feb. 24, 2026 (GLOBE NEWSWIRE) — Cardiff Oncology, Inc. (Nasdaq: CRDF), a clinical-stage biotechnology company leveraging PLK1 inhibition to develop novel therapies across a range of cancers, today announced financial results for the full year ended December 31, 2025, and provided a business update.
“Cardiff Oncology has entered 2026 with strong clinical momentum and a clear path for advancing onvansertib, our lead program, in first-line RAS-mutated metastatic colorectal cancer,” said Mani Mohindru, PhD, interim Chief Executive Officer. “Our focus in 2025 was on rigorous clinical execution, which allowed us to generate increasingly compelling evidence supporting onvansertib’s potential to improve patient outcomes in RAS-mutated mCRC, culminating in the latest positive data cut announced earlier this year. The CRDF-004 trial demonstrated a consistent, dose-dependent treatment benefit when onvansertib was added to FOLFIRI/bev, including a near 30% improvement in response rate over the control arm and encouraging durability trends as measured by progression-free survival. These data are in line with what we had previously seen in our second-line trial in bev-naive patients treated with onvansertib + FOLFIRI/bev. Given that it has been over two decades since there has been meaningful innovation for this patient population, we believe these results represent a transformative step forward.”
Continued Dr. Mohindru, “Based on these results, we plan to advance the 30 mg dose of onvansertib with FOLFIRI/bev into our proposed registrational program and expect to provide detailed data and registrational plans after discussions with the FDA in the first half of 2026. As we transition into late-stage clinical development and continue to strengthen our leadership and operational teams, we remain focused on disciplined execution, progressing our lead program toward a potential new standard of care in first-line RAS-mutated mCRC.”
Company highlights for the quarter ended December 31, 2025, and subsequent weeks
Positive update from randomized Phase 2 CRDF-004 trial in first-line RAS-mutated metastatic colorectal cancer (“mCRC”) support advancement of the onvansertib program into registrational development
In January 2026, Cardiff reported a positive update from CRDF-004, a randomized Phase 2 trial evaluating onvansertib in combination with standard of care (“SoC”) regimens in patients with first-line RAS-mutated mCRC. As of the January 22, 2026 cutoff in the intent-to-treat population, the 30 mg onvansertib + FOLFIRI/bev arm achieved a confirmed objective response rate (“ORR”) of 72.2%, compared to 43.2% across the combined SoC arms. The 30 mg onvansertib dose in combination with FOLFIRI/bev also demonstrated marked improvement in progression-free survival (“PFS”) versus FOLFIRI/bev (HR: 0.38) and combined SoC of FOLFOX/bev and FOLFIRI/bev (HR: 0.37, p<0.05), with no significant added toxicity observed.
Based on these results, the Company expects to advance the 30 mg dose of onvansertib in combination with FOLFIRI/bev into planned registrational development. Cardiff expects to share detailed Phase 2 CRDF-004 data and, after discussions with the FDA, provide registrational plans for onvansertib in combination with FOLFIRI/bev in first-line RAS-mutated mCRC in the first half of 2026.
Executive leadership team transitioned to support late-stage development
In January 2026, Cardiff announced executive leadership changes to support the Company’s transition into late-stage clinical development and advancement toward key clinical and corporate milestones. Mani Mohindru, PhD, a member of Cardiff’s Board of Directors since 2021 and an experienced biotechnology executive, was appointed interim Chief Executive Officer. Brigitte Lindsay was promoted to Chief Accounting Officer, ensuring continuity within the Company’s finance function. The Company has initiated a search for a permanent Chief Executive Officer and Chief Financial Officer.
Presentation of investigator-sponsored clinical data in chronic myelomonocytic leukemia (“CMML”) at the American Society of Hematology (“ASH”) Annual Meeting
In December 2025, clinical data from an investigator-sponsored Phase 1 trial evaluating onvansertib monotherapy in CMML were presented at ASH 2025. In the dose-escalation trial (N=9), onvansertib was generally well-tolerated and demonstrated preliminary efficacy in approximately 40% of patients, including one patient achieving an optimal bone marrow response. These clinical findings further validate onvansertib’s potential activity across both hematologic and solid tumors.
Full Year 2025 Financial Results
Liquidity and Cash Runway
As of December 31, 2025, Cardiff Oncology had approximately $58.3 million in cash, cash equivalents, and short-term investments. Based on its current operating and clinical plans and projected expenditures, the Company believes that its existing cash resources are sufficient to fund operations into the first quarter of 2027.
Operating Results
Total operating expenses for the year ended December 31, 2025 were approximately $49.6 million, compared to $49.3 million for the year ended December 31, 2024. The $0.3 million increase was primarily attributable to an increase in SG&A expense, driven mainly by strategic advisory services and incremental employee separation costs recorded in the current period. This increase was partially offset by lower R&D expenses related to clinical trial activity and outside services. The reduction in R&D expenses was partially offset by an increase in stock-based compensation expense during the current period.
About Cardiff Oncology, Inc. Cardiff Oncology is a clinical-stage biotechnology company advancing innovative cancer treatments focused on PLK1 inhibition, a validated oncology target with practice-changing potential. Our lead asset, onvansertib, is a highly specific, oral PLK1 inhibitor currently being evaluated in a Phase 2 trial for first-line treatment of RAS-mutated metastatic colorectal cancer (“mCRC”), addressing a large, underserved patient population with high unmet need. Onvansertib is also under investigation in other PLK1-driven cancers through ongoing investigator-initiated trials and has shown robust single agent clinical activity in hard-to-treat tumors. By targeting tumor vulnerabilities, we aim to overcome treatment resistance and deliver improved clinical outcomes for patients.
Forward-Looking Statements Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified using words such as “anticipate,” “believe,” “forecast,” “estimated” and “intend” or other similar terms or expressions that concern Cardiff Oncology’s expectations, strategy, plans or intentions. These forward-looking statements are based on Cardiff Oncology’s current expectations and actual results could differ materially. There are several factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results; our clinical trials may be suspended or discontinued due to unexpected side effects or other safety risks that could preclude approval of our product candidate; results of preclinical studies or clinical trials for our product candidate could be unfavorable or delayed; our need for additional financing; risks related to business interruptions, including the outbreak of COVID-19 coronavirus and cyber-attacks on our information technology infrastructure, which could seriously harm our financial condition and increase our costs and expenses; uncertainties of government or third party payer reimbursement; dependence on key personnel; limited experience in marketing and sales; substantial competition; uncertainties of patent protection and litigation; dependence upon third parties; and risks related to failure to obtain FDA clearances or approvals and noncompliance with FDA regulations. There are no guarantees that our product candidate will be utilized or prove to be commercially successful. Additionally, there are no guarantees that future clinical trials will be completed or successful or that our product candidate will receive regulatory approval for any indication or prove to be commercially successful. Investors should read the risk factors set forth in Cardiff Oncology’s Form 10-K for the year ended December 31, 2025, and other periodic reports filed with the Securities and Exchange Commission. While the list of factors presented here is considered representative, no such list should be considered to be a complete statement of all potential risks and uncertainties. Unlisted factors may present significant additional obstacles to the realization of forward-looking statements. Forward-looking statements included herein are made as of the date hereof, and Cardiff Oncology does not undertake any obligation to update publicly such statements to reflect subsequent events or circumstances.
FORT WORTH, Texas, Feb. 24, 2026 (GLOBE NEWSWIRE) — Sports Entertainment Gaming Global Corporation (NASDAQ: SEGG, LTRYW) (the “Company” or “SEGG Media”), the global sports, entertainment, and gaming group, today announced that Veloce Media Group (“Veloce”) co-founder and Quadrant CEO Jamie MacLaurin was appointed to the role of Senior Vice President of SEGG’s sports business. MacLaurin has been key in building one of the industry’s most dynamic motorsport businesses, spanning apparel, athletes, content and partnerships. In his new role, MacLaurin will not only continue to play a leading role in Veloce and Quadrant, he will also identify commercial opportunities for the benefit of SEGG Media’s sports business that compliment and enhance the Veloce and Quadrant business models.
As a result of completing the remaining tranches and additional share purchases, SEGG Media has now secured supermajority control of approximately 68% of Veloce’s issued and outstanding equity. To further streamline governance and align long-term strategic objectives, SEGG Media is extending a global offer to acquire the remaining minority equity interests in Veloce. The Company believes increased ownership will enhance operational efficiency, simplify the capital structure, and support growth initiatives across the combined platform. Veloce is expected to contribute $20 million in annual revenue, which SEGG will begin to recognize and report in Q1 of 2026, representing a material increase to SEGG’s consolidated top line.
Quadrant, which MacLaurin co-founded alongside 2025 Formula One World Champion Lando Norris, combines competitive racing with creator culture and lifestyle branding. Together, they have driven a successful diversification of the business model from gaming to content and lifestyle around motorsport, securing larger blue-chip partnerships with companies such as Electronic Arts (EA), VISA, LEGO and E.ON, as well as significantly increasing revenues. Veloce Media Group reported a 153% year-over-year increase in revenue between 2023 and 2024.
Under MacLaurin’s stewardship, Quadrant has grown exponentially. The brand now has an audience of nearly 7 million followers, directly contributing to a wider Veloce digital ecosystem that generates over 500 million monthly views and drives substantial yearly financial growth. Veloce acquired Quadrant in July 2025.
Robert Stubblefield, Chief Financial Officer and Interim Chief Executive Officer and President of SEGG Media, stated:“The acquisition of a supermajority interest in Veloce materially strengthens our revenue base and positions us to consolidate a high-growth international media platform. Appointing Jamie as SVP of SEGG’s sports business ensures continuity of leadership and operational execution as we focus on disciplined growth, capital efficiency, and scalable monetization across our business units.”
Mr. MacLaurin added: “Taking a leadership position at SEGG Media at such a transformative time is really exciting for me. The Company’s portfolio of digital assets gives us the ultimate platform to scale our vision globally, bridging the gap between creator-led culture and top-tier sports entertainment in completely innovative ways. With the Company’s access to capital markets and its global asset portfolio, we are positioned to accelerate Veloce’s growth trajectory and expand our commercial footprint.”
Prior to his ventures with Veloce and Quadrant, MacLaurin began his career as a sports agent, representing elite international athletes.
About SEGG Media
Sports Entertainment Gaming Global Corporation (Nasdaq: SEGG, LTRYW) is a global sports, entertainment, and gaming group operating a portfolio of digital and experiential assets including Sports.com, Concerts.com, TicketStub.com, Lottery.com, and Veloce Media Group. Through its expanding ecosystem of media, live experiences, gaming platforms, and creator-led content, the Company connects global audiences to the sports, events, and interactive entertainment they love. Focused on disciplined execution, ethical gaming, and scalable revenue generation, SEGG Media is building an integrated platform designed to drive sustainable growth and long-term shareholder value.
Important Notice Regarding Forward-Looking Statements
This press release contains statements that constitute “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements, other than statements of present or historical fact included in this press release, regarding the Company’s strategy, future operations, prospects, plans and objectives of management, are forward-looking statements. The words “could,” “should,” “will,” “may,” “believe,” “anticipate,” “intend,” “estimate,” “expect,” “project,” “initiatives,” “continue,” the negative of such terms and other similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain such identifying words. These forward-looking statements are based on management’s current expectations and assumptions about future events and are based on currently available information as to the outcome and timing of future events. The forward-looking statements speak only as of the date of this press release or as of the date they are made. The Company cautions you that these forward-looking statements are subject to numerous risks and uncertainties, most of which are difficult to predict and many of which are beyond the control of the Company. In addition, the Company cautions you that the forward-looking statements contained in this press release are subject to risks and uncertainties, including but not limited to, any future findings from ongoing review of the Company’s internal accounting controls, additional examination of the preliminary conclusions of such review, the Company’s ability to secure additional capital resources, the Company’s ability to continue as a going concern, the Company’s ability to respond in a timely and satisfactory matter to the inquiries by Nasdaq, the Company’s ability to regain compliance with the Bid Price Requirement, the Company’s ability to regain compliance with Nasdaq Listing Rules, the Company’s ability to become current with its SEC reports, and those additional risks and uncertainties discussed under the heading “Risk Factors” in the Form 10-K/A filed by the Company with the SEC on April 22, 2025, and the other documents filed, or to be filed, by the Company with the SEC. Additional information concerning these and other factors that may impact the operations and projections discussed herein can be found in the reports that the Company has filed and will file from time to time with the SEC. These SEC filings are available publicly on the SEC’s website at www.sec.gov. Should one or more of the risks or uncertainties described in this press release materialize or should underlying assumptions prove incorrect, actual results and plans could differ materially from those expressed in any forward-looking statements. Except as otherwise required by applicable law, the Company disclaims any duty to update any forward-looking statements, all of which are expressly qualified by the statements in this section, to reflect events or circumstances after the date of this press release.
This press release was published by a CLEAR® Verified individual.
For additional information, visit www.seggmediacorp.com or contact media relations at media@seggmediacorp.com.
Members Bring Deep Expertise in Immuno-Oncology, Translational Medicine, and Checkpoint Inhibitor Combination Strategies
ATLANTA, GA – February 24, 2026 – GeoVax Labs, Inc. (Nasdaq: GOVX), a clinical-stage biotechnology company developing immunotherapies and vaccines for cancers and infectious diseases, today announced the formation of its Oncology Advisory Board with the appointment of three internationally recognized leaders in immuno-oncology, translational medicine, and clinical development.
This Advisory Board will play a central role in guiding the scientific, translational, and clinical advancement of GeoVax’s oncology program, focused primarily on Gedeptin®, the company’s gene-directed enzyme prodrug therapeutic (GDEPT). GeoVax plans to conduct a Phase 2 trial with Gedeptin in the neoadjuvant setting, pairing it with an immune checkpoint inhibitor (ICI) in locally advanced head and neck squamous cell carcinoma. In parallel, it will be evaluating combination Gedeptin + ICI strategies across additional solid tumor indications.
Oncology Advisory Board Members
Chas Bountra, PhD, OBE Professor of Translational Medicine, University of Oxford Former Pro-Vice-Chancellor for Innovation, University of Oxford Former Vice President & Head of Biology, GlaxoSmithKline
Dr. Bountra brings extensive leadership spanning academia, biotech, and global pharmaceutical R&D. He is widely recognized for building translational drug discovery engines, advancing novel therapeutic modalities into the clinic, and creating high-impact academic–industry partnerships. His expertise in translational medicine, drug development strategy, and innovation ecosystems will support Gedeptin’s progression from proof-of-concept to clinically scalable development programs aligned with regulatory and partnership pathways.
Marc S. Ernstoff, MD Director, Experimental Cell Therapy, Dartmouth Health Former Chief, Immuno-Oncology Branch, National Cancer Institute (NIH)
Dr. Ernstoff is a recognized pioneer in cancer immunotherapy with significant experience across cytokine therapy, checkpoint inhibitors, and combination immuno-oncology trials. His career includes leadership roles at the National Cancer Institute, Roswell Park Comprehensive Cancer Center, Dartmouth-Hitchcock, and Yale, with direct involvement in numerous landmark immunotherapy studies. His expertise will inform Gedeptin’s clinical positioning, trial design considerations, and biomarker-driven evaluation alongside immune checkpoint inhibitors, as well as its potential role in immune-sensitizing solid tumors.
Anthony J. Olszanski, MD, RPh Professor of Medicine and Vice Chair for Clinical Research Fox Chase Cancer Center
Dr. Olszanski is a nationally recognized leader in early-phase oncology drug development, with deep experience running first-in-human and Phase 1/2 trials across solid tumors, including multiple checkpoint inhibitor combinations, oncolytic approaches, and novel immune-modulating agents. His background in clinical pharmacology and trial execution will be instrumental as GeoVax advances Gedeptin into combination, neoadjuvant, and expansion-stage clinical settings.
Strategic Focus: Advancing Gedeptin in Combination Immuno-Oncology
This Oncology Advisory Board, with expertise in oncology and translational medicine, reflects GeoVax’s increasing focus on Gedeptin as a differentiated immune-sensitizing platform, particularly in combination with immune checkpoint inhibitors. Gedeptin’s intratumoral delivery and localized tumor-debulking mechanism are designed to enhance antigen release and immune activation within the tumor microenvironment – an approach that may complement and extend the efficacy of systemic checkpoint blockade.
In addition to scientific strategy, these advisors will provide integrated guidance on clinical trial design, translational biomarker strategy, patient selection, and regulatory-aligned development pathways as Gedeptin advances through combination and neoadjuvant clinical programs.
David A. Dodd, Chairman & Chief Executive Officer of GeoVax, commented: “The addition of these three exceptional oncology leaders significantly strengthens our scientific and clinical foundation as we advance Gedeptin into its next stage of development. Their collective experience – from early drug discovery through late-stage immuno-oncology trials and regulatory strategy – directly aligns with our goal of positioning Gedeptin as a novel immune-sensitizing therapy across solid tumors.”
Kelly T. McKee, MD, Chief Medical Officer of GeoVax, added: “Gedeptin sits at the intersection of localized tumor control and systemic immune activation. As checkpoint inhibitors move earlier in treatment paradigms, including neoadjuvant settings, expert guidance on trial design, patient selection, and translational endpoints is critical as we move into clinically consequential combination strategies. We anticipate that this Advisory Board will provide precisely that level of integrated scientific and clinical insight.”
About Gedeptin®
Gedeptin® is a gene-directed enzyme prodrug therapy (GDEPT) delivered intratumorally using a non-replicating viral vector encoding purine nucleoside phosphorylase (PNP). Following systemic administration of a prodrug, the encoded enzyme converts it into a cytotoxic agent directly within the tumor microenvironment, selectively destroying tumor cells while promoting immune recognition. Gedeptin has completed a multicenter Phase 1/2 clinical trial in advanced head and neck cancer and has received Orphan Drug Designation for oral and pharyngeal cancers.
About GeoVax
GeoVax Labs, Inc. is a clinical-stage biotechnology company focused on the development of vaccines and immunotherapies addressing high-consequence infectious diseases and solid tumor cancers. GeoVax’s priority program is GEO-MVA, a Modified Vaccinia Ankara (MVA)–based vaccine targeting mpox and smallpox. The program is advancing under an expedited regulatory pathway, with plans to initiate a pivotal Phase 3 clinical trial in the second half of 2026, to address critical global needs for expanded orthopoxvirus vaccine supply and biodefense preparedness. In oncology, GeoVax is developing Gedeptin®, a gene-directed enzyme prodrug therapy (GDEPT) designed to enhance immune checkpoint inhibitor activity. Gedeptin has completed a multicenter Phase 1/2 clinical trial in advanced head and neck cancer and is being advanced into combination strategies, including planned neoadjuvant and first-line settings. GeoVax’s broader pipeline includes the development of GEO-CM04S1, a next-generation COVID-19 vaccine candidate being evaluated in immunocompromised and other patient populations. GeoVax maintains a global intellectual property portfolio supporting its infectious disease and oncology programs and continues to evaluate strategic partnerships and funding opportunities aligned with its development priorities. For more information, visit www.geovax.com.
Forward-Looking Statements
This release contains forward-looking statements regarding GeoVax’s business plans. The words “believe,” “look forward to,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “could,” “target,” “potential,” “is likely,” “will,” “expect” and similar expressions, as they relate to us, are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. Actual results may differ materially from those included in these statements due to a variety of factors, including whether: GeoVax is able to obtain acceptable results from ongoing or future clinical trials of its investigational products, GeoVax’s immuno-oncology products and preventative vaccines can provoke the desired responses, and those products or vaccines can be used effectively, GeoVax’s viral vector technology adequately amplifies immune responses to cancer antigens, GeoVax can develop and manufacture its immuno-oncology products and preventative vaccines with the desired characteristics in a timely manner, GeoVax’s immuno-oncology products and preventative vaccines will be safe for human use, GeoVax’s vaccines will effectively prevent targeted infections in humans, GeoVax’s immuno-oncology products and preventative vaccines will receive regulatory approvals necessary to be licensed and marketed, GeoVax raises required capital to complete development, there is development of competitive products that may be more effective or easier to use than GeoVax’s products, GeoVax will be able to enter into favorable manufacturing and distribution agreements, and other factors, over which GeoVax has no control.
Further information on our risk factors is contained in our periodic reports on Form 10-Q and Form 10-K that we have filed and will file with the SEC. Any forward-looking statement made by us herein speaks only as of the date on which it is made. Factors or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of them. We undertake no obligation to publicly update any forward-looking statement, whether as a result of new information, future developments or otherwise, except as may be required by law.
Ongoing Phase 3 full approval clinical trial of ateganosine holds high probability of technical success for interim and full analysis
FDA Fast Track designation offers clear pathway for ateganosine as third-line therapy for non-small cell lung cancer (NSCLC)
First and only direct telomere-targeting anticancer agent in clinical development anywhere
MAIA CEO details development pipeline in letter to shareholders
CHICAGO, Feb. 24, 2026 (GLOBE NEWSWIRE) — MAIA Biotechnology, Inc. (NYSE American: MAIA) (“MAIA”, the “Company”), a clinical-stage biopharmaceutical company focused on developing targeted immunotherapies for cancer, today published a 2026 Letter to Shareholders by Founder and CEO Vlad Vitoc, M.D. featuring the Company’s strong momentum in clinical trials of its lead molecule, ateganosine, as a treatment for non-small cell lung cancer (NSCLC). As a potential breakthrough therapy, ateganosine holds substantial commercial opportunity in a $50 billion global immunotherapy market.1
As stated in the Letter, Dr. Vitoc wrote, “Our development strategy intentionally targets the third-line (3L) NSCLC population, where the unmet need is urgent. No established standard of care exists in 3L treatment, with most oncologists currently treating 3L patients with chemotherapy, leading to particularly poor clinical outcomes. In clinical studies, ateganosine sequenced with an immune checkpoint inhibitor (CPI) has demonstrated outcomes that exceed those historically achieved with either CPI-based therapy or chemotherapy alone. These findings position ateganosine not as a competitor to CPIs, but as the foundation of a new treatment category designed specifically for advanced NSCLC following CPI and chemotherapy failure. By focusing on a third-line population with no defined standard of care, we are addressing an underserved group of approximately 50,000 patients annually in the United States and creating a differentiated, incremental revenue opportunity outside of the CPI market.
“Ateganosine could mark the start of a new therapeutic category in cancer treatment and could become the standard of care for multiple cancer indications,” Dr. Vitoc added. “The commercial opportunity for ateganosine could be immense.”
Dr. Vitoc concluded his Letter with the following statement: “As we move forward, we are optimistic about the progress and potential outcomes of our advanced trials and the broader promise of ateganosine. We are grateful to our stockholders, employees, partners and investigators, for their continued support and commitment. With strong momentum and a clear path ahead, we believe MAIA Biotechnology’s future is bright and rich with opportunity.”
MAIA’s 2026 Letter to Shareholders is available in its entirety at ir.maiabiotech.com.
About Ateganosine
Ateganosine (THIO, 6-thio-dG or 6-thio-2’-deoxyguanosine) is a first-in-class investigational telomere-targeting agent currently in clinical development to evaluate its activity in non-small cell lung cancer (NSCLC). Telomeres, along with the enzyme telomerase, play a fundamental role in the survival of cancer cells and their resistance to current therapies. The modified nucleotide 6-thio-2’-deoxyguanosine induces telomerase-dependent telomeric DNA modification, DNA damage responses, and selective cancer cell death. Ateganosine-damaged telomeric fragments accumulate in cytosolic micronuclei and activates both innate (cGAS/STING) and adaptive (T-cell) immune responses. The sequential treatment of ateganosine followed by PD-(L)1 inhibitors resulted in profound and persistent tumor regression in advanced, in vivo cancer models by induction of cancer type–specific immune memory. Ateganosine is presently developed as a second or later line of treatment for NSCLC for patients that have progressed beyond the standard-of-care regimen of existing checkpoint inhibitors.
About MAIA Biotechnology, Inc.
MAIA is a targeted therapy, immuno-oncology company focused on the development and commercialization of potential first-in-class drugs with novel mechanisms of action that are intended to meaningfully improve and extend the lives of people with cancer. Our lead program is ateganosine (THIO), a potential first-in-class cancer telomere targeting agent in clinical development for the treatment of NSCLC patients with telomerase-positive cancer cells. For more information, please visit www.maiabiotech.com.
Forward Looking Statements
MAIA cautions that all statements, other than statements of historical facts contained in this press release, are forward-looking statements. Forward-looking statements are subject to known and unknown risks, uncertainties, and other factors that may cause our or our industry’s actual results, levels or activity, performance or achievements to be materially different from those anticipated by such statements. The use of words such as “may,” “might,” “will,” “should,” “could,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “project,” “intend,” “future,” “potential,” or “continue,” and other similar expressions are intended to identify forward looking statements. However, the absence of these words does not mean that statements are not forward-looking. For example, all statements we make regarding (i) the initiation, timing, cost, progress and results of our preclinical and clinical studies and our research and development programs, (ii) our ability to advance product candidates into, and successfully complete, clinical studies, (iii) the timing or likelihood of regulatory filings and approvals, (iv) our ability to develop, manufacture and commercialize our product candidates and to improve the manufacturing process, (v) the rate and degree of market acceptance of our product candidates, (vi) the size and growth potential of the markets for our product candidates and our ability to serve those markets, and (vii) our expectations regarding our ability to obtain and maintain intellectual property protection for our product candidates, are forward looking. All forward-looking statements are based on current estimates, assumptions and expectations by our management that, although we believe to be reasonable, are inherently uncertain. Any forward-looking statement expressing an expectation or belief as to future events is expressed in good faith and believed to be reasonable at the time such forward-looking statement is made. However, these statements are not guarantees of future events and are subject to risks and uncertainties and other factors beyond our control that may cause actual results to differ materially from those expressed in any forward-looking statement. Any forward-looking statement speaks only as of the date on which it was made. We undertake no obligation to publicly update or revise any forward-looking statement, whether as a result of new information, future events or otherwise, except as required by law. In this release, unless the context requires otherwise, “MAIA,” “Company,” “we,” “our,” and “us” refers to MAIA Biotechnology, Inc. and its subsidiaries.
Greater than 25% absolute reduction in thrombotic events with CAD-1005 versus placebo on a background of standard anticoagulant therapy, despite no difference in platelet count recovery
End-of-Phase 2 Meeting Scheduled for March 2026
PONTE VEDRA, Fla., Feb. 24, 2026 (GLOBE NEWSWIRE) — Cadrenal Therapeutics, Inc. (Nasdaq: CVKD), a late-stage biopharmaceutical company advancing novel therapies for life-threatening immune and thrombotic conditions, today announced encouraging results from a Phase 2 trial evaluating CAD-1005 (formerly VLX-1005) in patients with heparin-induced thrombocytopenia (HIT), a severe pro-thrombotic reaction to heparin, the most commonly used parenteral anticoagulant.
This randomized, blinded, placebo-controlled trial evaluated the safety and efficacy of CAD-1005, a selective inhibitor of 12-lipoxygenase (12-LOX), a critical immune signaling pathway implicated in HIT, in patients receiving standard anticoagulant therapy. To potentially validate a new surrogate endpoint, the previous investigational new drug sponsor, Veralox Therapeutics, selected platelet count recovery rate as the primary endpoint. Their trial did not meet this primary endpoint. The secondary endpoint was the incidence of new or worsening thrombotic events, including radiologic progression, which showed encouraging results. The study concluded in December 2025 following the transfer of program ownership from Veralox to Cadrenal. Although CAD-1005 did not significantly affect platelet recovery rate, CAD-1005-treated patients had fewer thrombotic events.
Highlights:
Primary Endpoint: Thrombotic events continued to occur even after platelet count recovery in both groups. Platelet recovery rates were similar between the CAD-1005 and placebo arms. Platelet count recovery did not appear to be a surrogate marker for clinical efficacy.
Key Secondary Endpoint: A high rate of thrombotic events (>75%) was observed in the placebo group, with fewer thrombotic events in the CAD-1005 group (50%), although the study was not powered to detect statistical significance. Adding an inhibitor of 12-LOX to standard anticoagulants to block the immunological mechanisms driving HIT may be more effective than anticoagulants alone in preventing thrombotic events.
Building on these secondary endpoint results, Cadrenal has been granted an End-of-Phase 2 (EOP2) meeting with the U.S. Food and Drug Administration (FDA) to align on a Phase 3 registration path. The Company considers this meeting a significant milestone in the development of CAD-1005, the only 12-LOX inhibitor in clinical development worldwide.
“The encouraging trend toward reduced thrombotic events in the CAD-1005 treatment arm is strong support for the company’s decision to acquire this asset and rapidly progress its development,” said Quang X. Pham, CEO of Cadrenal Therapeutics. “Inhibition of 12-LOX is an exciting therapeutic frontier, potentially targeting numerous inflammatory, thrombotic, and metabolic conditions.”
“We learned two very important things from this study, the only blinded placebo-controlled trial ever conducted in HIT,” said James Ferguson, MD, Chief Medical Officer of Cadrenal Therapeutics. “First, platelet count recovery was not an appropriate surrogate endpoint for clinical efficacy in a trial in which standard therapy event rates were strikingly high. Secondly, despite the relatively small number of patients, the reduction in thrombotic events with CAD-1005 is extremely encouraging. CAD-1005 could represent a major step forward as the only first-line therapy targeting the immune mechanisms responsible for HIT.”
“Our field (HIT) is full of anticoagulant use in the absence of randomized prospective trials,” said Steven E. McKenzie, MD, PhD, Professor of Medicine at Thomas Jefferson University and a member of the study steering committee. “We are enthusiastic about CAD-1005 in addressing both the underlying immune mechanism and the unmet medical need for this serious thrombotic disorder.”
Detailed trial results will be presented at a future scientific meeting.
About Heparin-Induced Thrombocytopenia (HIT)
Heparin is the most widely used in-hospital anticoagulant, with over 12 million patients receiving it in the United States each year. Heparin-induced thrombocytopenia (HIT) is a potentially life-threatening immune-mediated complication of heparin administration that occurs when antibodies to heparin activate platelets, leading to clots throughout the circulatory system, dramatically lowering platelet counts, and increasing the risk of bleeding. Complications of HIT include deep vein thrombosis, pulmonary embolism, stroke, myocardial infarction, amputation, and death, with mortality rates for HIT exceeding 20% in some studies. CAD-1005 is the only treatment in clinical development that targets the underlying immune drivers of HIT.
About CAD-1005
CAD-1005 is an investigational therapy being evaluated for the treatment of suspected HIT. CAD-1005 is designed to selectively inhibit 12-LOX, a pathway integral to the primary immune mechanisms driving HIT. Unlike existing therapies for HIT, which are only directed at preventing thrombotic complications, this approach addresses the primary underlying cause of HIT. In preclinical models of HIT, CAD-1005 has been shown to prevent or treat HIT and halt the development of both thrombocytopenia and blood clots. The drug has not been associated with increased bleeding in animals or healthy human volunteers. CAD-1005 has received Orphan Drug Designation (ODD) and Fast Track designation from the U.S. Food and Drug Administration, as well as orphan drug status from the European Medicines Agency.
About the Study
The study was originally planned to enroll 60 patients, but was stopped in December 2025 after program ownership transferred to Cadrenal. Analysis of all existing trial data was recently completed. The final dataset includes 24 patients with a presumptive diagnosis of HIT, randomized to receive either CAD-1005 or a matching placebo; all patients received concomitant standard anticoagulant therapy, either argatroban or bivalirudin. The primary endpoint was the rate of platelet count recovery; a key secondary endpoint was the development of new or worsening thrombotic events, the composite of death, stroke, systemic embolism, myocardial infarction, deep venous thrombosis, superficial vein thrombosis, or skin necrosis. Primary analyses focused on 17 patients in whom HIT was confirmed by a central lab functional assay.
About Cadrenal Therapeutics, Inc.
Cadrenal Therapeutics, Inc. (Nasdaq: CVKD) is a late-stage biopharmaceutical company advancing novel therapies for life-threatening immune and thrombotic conditions. Its lead program, CAD-1005, is a first-in-class 12-LOX inhibitor for the treatment of heparin-induced thrombocytopenia (HIT), a deadly immune-mediated thrombotic disorder. CAD-1005 has received Orphan Drug and Fast Track designations from the U.S. Food and Drug Administration, as well as orphan drug status from the European Medicines Agency. Second-generation 12-LOX oral therapeutics are also under development.
The Company’s broader pipeline includes tecarfarin, a Phase 3-ready oral vitamin K antagonist for the treatment of patients with end-stage kidney disease and those with left ventricular assist devices, and frunexian, a parenteral, clinical-stage Factor XIa inhibitor designed for use in acute hospital settings. For more information, visit https://www.cadrenal.com/ and connect with the Company on LinkedIn.
Safe Harbor
Any statements in this press release about future expectations, plans, and prospects, as well as any other statements regarding matters that are not historical facts, may constitute “forward-looking statements.” The words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potentially,” “predict,” “project,” “should,” “target,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. These statements include adding an inhibitor of 12-LOX to standard anticoagulants to block the immunological mechanisms driving HIT being more effective than anticoagulants alone in preventing thrombotic events; the encouraging trend toward reduced thrombotic events in the CAD-1005 treatment arm being strong support for the Cadrenal’s decision to acquire this asset and rapidly progress its clinical development; full trial results being presented at a future scientific meeting; the reduction in thrombotic events with CAD-1005 being extremely encouraging, despite the relatively small number of patients; CAD-1005 representing a major step forward as the only first-line therapy targeting the immune mechanisms responsible for HIT; The EOP2 meeting being a significant milestone in the development of CAD-1005; CAD-1005 addressing both the underlying immune mechanism and the unmet medical need for this serious thrombotic disorder; the encouraging trend toward reduced thrombotic events in the CAD-1005 treatment arm being strong support for Cadrenal’s decision to acquire this asset and rapidly progress its development; CAD -1005 addressing the underlying immune drivers of HIT; and presenting detailed trial results at a future scientific meeting. Actual results may differ materially from those indicated by such forward-looking statements as a result of various important factors, including Cadrenal’s ability to advance the clinical development of CAD-1005 for the treatment of HIT, including designing a pivotal Phase 3 registration study acceptable to the FDA; CAD-1005 having the ability to address the underlying immune mechanism and the unmet medical need for the serious thrombotic disorder; Cadrenal’s ability to continue to advance novel therapeutics to treat or prevent thrombosis in high-risk patients; Cadrenal’s ability to successfully complete clinical trials on time and achieve desired results and benefits as expected including support for CAD-1005’s potential to be a treatment option for HIT, Cadrenal’s ability to obtain regulatory approvals for commercialization of product candidates or to comply with ongoing regulatory requirements and the other risk factors described in the Company’s Annual Report on Form 10-K for the year ended December 31, 2024, and the Company’s subsequent filings with the Securities and Exchange Commission, including subsequent periodic reports on Quarterly Reports on Form 10-Q and Current Reports on Form 8-K. Any forward-looking statements contained in this press release speak only as of the date hereof and, except as required by federal securities laws, the Company specifically disclaims any obligation to update any forward-looking statement, whether as a result of new information, future events, or otherwise.
Company strengthens industry-leading compliance program with new per-transaction identity verification procedure, enhancing protections against Crypto ATM fraud
ATLANTA, Feb. 24, 2026 (GLOBE NEWSWIRE) — Bitcoin Depot (NASDAQ: BTM), a U.S.-based Bitcoin ATM (“BTM”) operator and leading fintech company, today announced it has initiated a phased rollout of a new compliance enhancement requiring customers to provide identification for every transaction at its kiosks, strengthening the Company’s safeguards against potential misuse. Bitcoin Depot is the first major operator in the industry to implement per-transaction ID collection, representing a significant advancement in its compliance protocols and ongoing efforts to prevent fraud and other illicit activity.
The policy began rolling out in February 2026 and is being implemented across Bitcoin Depot’s U.S. kiosk network, further strengthening the Company’s Know Your Customer (“KYC”) standards. By requiring identification for every transaction, the enhancement adds another layer of protection designed to help prevent account sharing, identity theft, and account takeover attempts as deployment continues.
“Bitcoin Depot has always prioritized compliance and consumer protection, and it’s crucial to our operations that we demonstrate proactive leadership in preventing fraud and building trust with our customers,” said Bitcoin Depot CEO Scott Buchanan. “Continuous verification allows us to detect suspicious activity based on customers, locations, or transaction amount before a transaction is approved. By requiring identity verification at every transaction, we are taking an additional step to strengthen security, protect customers, and maintain the integrity of our services.”
Building on its previously announced “First-Transaction ID Verification” policy, the new requirement extends identity checks beyond initial onboarding, introducing an additional layer of oversight for returning users. This approach enables Bitcoin Depot to apply a higher standard of transaction monitoring and to detect suspicious or unauthorized activity in real time.
Bitcoin Depot believes the updated compliance measure will offer meaningful protection for consumers as the digital asset industry continues to mature and works to eliminate misuse by bad actors.
Bitcoin Depot’s kiosks allow customers to seamlessly convert cash into Bitcoin, which customers can use to access the broader digital financial system for payments, transfers, remittances, and investments. Since becoming the first U.S. Bitcoin ATM operator to go public in July 2023, Bitcoin Depot has demonstrated its ability to expand domestically and internationally while maintaining a focus on compliance, access, and customer experience.
About Bitcoin Depot Bitcoin Depot Inc. (Nasdaq: BTM) was founded in 2016 with the mission to connect those who prefer to use cash to the broader, digital financial system. Bitcoin Depot provides its users with simple, efficient and intuitive means of converting cash into Bitcoin, which users can deploy in the payments, spending and investing space. Users can convert cash to bitcoin at Bitcoin Depot kiosks in 47 states and at thousands of name-brand retail locations in 31 states through its BDCheckout product. The Company has the largest market share in North America and operates over 9,000 kiosk locations globally as of August 2025. Learn more at www.bitcoindepot.com.
Cautionary Note Regarding Forward-Looking Statements This press release and any oral statements made in connection herewith include “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Exchange Act. Forward-looking statements are any statements other than statements of historical fact, and include, but are not limited to, statements regarding the expectations of plans, business strategies, objectives and growth and anticipated financial and operational performance, including our growth strategy and ability to increase deployment of our products and services, the anticipated effects of the Agreement. These forward-looking statements are based on management’s current beliefs, based on currently available information, as to the outcome and timing of future events. Forward-looking statements are often identified by words such as “anticipate,” “appears,” “approximately,” “believe,” “continue,” “could,” “designed,” “effect,” “estimate,” “evaluate,” “expect,” “forecast,” “goal,” “initiative,” “intend,” “may,” “objective,” “outlook,” “plan,” “potential,” “priorities,” “project,” “pursue,” “seek,” “should,” “target,” “when,” “will,” “would,” or the negative of any of those words or similar expressions that predict or indicate future events or trends or that are not statements of historical matters, although not all forward-looking statements contain such identifying words. In making these statements, we rely upon assumptions and analysis based on our experience and perception of historical trends, current conditions, and expected future developments, as well as other factors we consider appropriate under the circumstances. We believe these judgments are reasonable, but these statements are not guarantees of any future events or financial results. These forward-looking statements are provided for illustrative purposes only and are not intended to serve as, and must not be relied on by any investor as, a guarantee, an assurance, a prediction or a definitive statement of fact or probability. Actual events and circumstances are difficult or impossible to predict and will differ from assumptions. Many actual events and circumstances are beyond our control.
These forward-looking statements are subject to a number of risks and uncertainties, including changes in domestic and foreign business, market, financial, political and legal conditions; failure to realize the anticipated benefits of the business combination; future global, regional or local economic and market conditions; the development, effects and enforcement of laws and regulations; our ability to manage future growth; our ability to develop new products and services, bring them to market in a timely manner and make enhancements to our platform; the effects of competition on our future business; our ability to issue equity or equity-linked securities; the outcome of any potential litigation, government and regulatory proceedings, investigations and inquiries; and those factors described or referenced in filings with the Securities and Exchange Commission. If any of these risks materialize or our assumptions prove incorrect, actual results could differ materially from the results implied by these forward-looking statements. There may be additional risks that we do not presently know or that we currently believe are immaterial that could also cause actual results to differ from those contained in the forward-looking statements. In addition, forward-looking statements reflect our expectations, plans or forecasts of future events and views as of the date of this press release. We anticipate that subsequent events and developments will cause our assessments to change.
We caution readers not to place undue reliance on forward-looking statements. Forward-looking statements speak only as of the date they are made, and we undertake no obligation to update publicly or otherwise revise any forward-looking statements, whether as a result of new information, future events, or other factors that affect the subject of these statements, except where we are expressly required to do so by law. All written and oral forward-looking statements attributable to us are expressly qualified in their entirety by this cautionary statement.
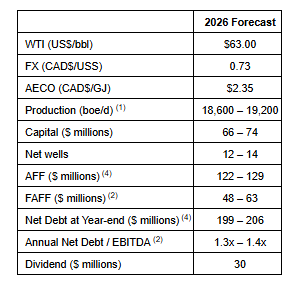
CALGARY AB, Feb. 24, 2026 /CNW/ – InPlay Oil Corp. (TSX: IPO) (OTCQX: IPOOF) (“InPlay” or the “Company”) announces that its Board of Directors have approved a capital program of $66 – $74 million for 2026.
InPlay had a stellar 2025 with an accretive and transformational acquisition in our core area and a very successful drilling program. Throughout 2025, InPlay delivered improved capital efficiencies through the successful application of enhanced drilling and completion techniques, driving production results that exceeded internally modelled type curves while achieving well costs below budget. InPlay’s improved capital efficiencies allowed the Company to increase its production guidance three times during 2025 with reduced capital spending.
InPlay’s 2026 capital budget reflects a disciplined and capital efficient program focused on strong production growth, maximizing Free Adjusted Funds Flow (“FAFF”)(2) and debt reduction. The Company plans to drill 12 – 14 net horizontal Cardium wells during 2026, with the majority of capital directed toward its Cardium-focused light oil assets in Pembina. InPlay’s 2026 capital budget reflects the improved capital efficiencies realized in 2025.
Key highlights of the 2026 capital program include:
Production Growth:
Forecasted average annual production of 18,600 – 19,200 boe/d(1) (60% – 62% light oil and NGLs), an 11% increase (based on mid-point) compared to estimated 2025, driven by:
Low corporate base decline rate of 22% due to the favorable decline profile;
Strong corporate netbacks driven by high oil and liquids weighting; and
Enhanced capital efficiencies from high graded drilling inventory.
FAFF Generation and Dividend Sustainability:
AFF(2) of $122 – $129 million;
FAFF of $48 – $63 million equating to a 11% – 15% FAFF Yield(3). FAFF exceeds the base annual dividend of $30 million (based on the current monthly dividend rate of $0.09/share or $1.08/share annualized) insulating the Company in the event of commodity price fluctuations.
InPlay’s dividend represents a dividend yield of approximately 7.0% at the current share price.
Debt Reduction:
Excess FAFF(3) is planned to be used to reduce debt;
Year-end Net Debt(2) of $199 – $206 million.
InPlay currently has forecasted commodity pricing similar to peers who have previously released 2026 guidance. To mitigate downside risk, InPlay has implemented a comprehensive hedging program providing protection against current market volatility. Details of the Company’s current hedges are provided in the “Hedging Summary” section of the Reader Advisories.
The table below outlines InPlay’s 2026 guidance:
In the first quarter of 2026, the Company plans to have its most active capital spend quarter of the year with five (5.0 net) horizontal wells being drilled. To date, InPlay has drilled and recently completed a two (2.0 net) ERH well-pad which have recently come on production. InPlay has also started drilling operations on a three (3.0 net) ERH well-pad which is expected to come on-line at the end of March. The majority of the capital spend on the remaining 7 – 9 net horizontal wells planned for the year is expected to occur in the second half of 2026.
InPlay continues to closely monitor global trade, geopolitical and commodity dynamics, proactively evaluating capital plans in response to pricing volatility, inflationary cost pressures, and other factors affecting the business. The Company will remain flexible and make decisions based on our core strategy of disciplined capital allocation, maintaining financial strength to ensure the long term sustainability of our strategy and return to shareholder program. Should commodity prices improve and stabilize, the Company will remain disciplined and flexible, with the ability to swiftly adjust its capital activity to align with evolving market conditions.
2025 Update
The Company is finalizing its results for 2025 and expects to achieve production of approximately 17,000 boe/d(1) (61% light crude oil and liquids) in line with the mid-point of our last forecast of 16,900 – 17,100 boe/d and 600 boe/d ahead of the mid-point of our original post acquisition forecast of 16,000 – 16,800 boe/d. In comparison to average production of 8,712 boe/d in 2024, production increased by approximately 95% in 2025.
Looking ahead after a transformation year with efficient capital spending, we remain focused on continued profitable development of our high-return asset base and are committed to delivering strong returns to shareholders through 2026 and beyond. On behalf of the management team and Board of Directors, we extend our gratitude to our employees, shareholders and bondholders for their support of the Company and the Canadian oil and gas industry.
NEW YORK–(BUSINESS WIRE)– Perfect Corp. (NYSE: PERF) (“Perfect” or the “Company”), a leading artificial intelligence (“AI”) company offering AI and augmented reality (“AR”) powered solutions to beauty, fashion, photo and video creative industries, today announced its unaudited financial results for the three months and the full year ended December 31, 2025.
Financial Results for the Three Months Ended December 31, 2025
Revenue
Total revenue was $18.1 million for the three months ended December 31, 2025, compared to $15.9 million in the same period of 2024, an increase of 14.2%. The increase was primarily due to strong growth momentum in the revenue of mobile app and web services subscriptions.
AI- and AR- cloud solutions and subscription revenue was $16.4 million for the three months ended December 31, 2025, compared to $15.1 million in the same period of 2024, an increase of 8.7%. The increase was driven by the continued revenue growth of YouCam mobile apps and web services subscriptions, the continued popularity among consumers of Generative AI technologies and AI editing features for photos and videos, and the stable demand for the Company’s online virtual product try-on solutions from brand customers.
Licensing revenue was $0.6 million for the three months ended December 31, 2025, compared to $0.5 million in the same period of 2024, an increase of 8.0%.
Others revenue was $1.2 million for the three months ended December 31, 2025, compared to $0.3 million in the same period of 2024, an increase of 286.1%. The increase was driven by the growth of virtual points purchased and consumed by end users. Virtual points are used for AI-powered services available on YouCam mobile apps and web services.
Gross Profit
Gross profit was $14.6 million for the three months ended December 31, 2025, compared with $11.8 million in the same period of 2024, an increase of 24.1%. Gross margin was 80.5% for the three months ended December 31, 2025, up from 74.1% in the same period of 2024. The increase in gross margin during this quarter was primarily due to the increased operational efficiency resulting from the ongoing realignment of engineering professionals as we continue to transition from customization of software toward more standardized AI solutions for our customer base.
Total Operating Expenses
Total operating expenses were $15.2 million for the three months ended December 31, 2025, compared with $12.2 million in the same period of 2024, an increase of 24.1%. The increase was primarily due to increases in research and development expenses and sales and marketing expenses, which were partially offset by a decrease in general and administrative expenses in the fourth quarter of 2025.
Sales and marketing expenses were $7.7 million for the three months ended December 31, 2025, compared to $6.9 million during the same period of 2024, an increase of 11.4%. This increase was primarily due to an increase in marketing events and advertising expenses related to our mobile apps and web services subscription.
Research and development expenses were $3.9 million for the three months ended December 31, 2025, compared to $2.8 million during the same period of 2024, an increase of 39.6%. The increase was primarily due to an increase in R&D headcount and related personnel costs including those arising from the acquisition of Wannaby Inc. (“Wannaby”), which was completed in January 2025.
General and administrative expenses were $1.5 million for the three months ended December 31, 2025, compared to $1.8 million during the same period of 2024, a decrease of 11.9%. The decrease was primarily due to reduced corporate insurance premium and external professional service fees.
Impairment loss on goodwill was $2.0 million for the three months ended December 31, 2025. No such impairment was recorded in the same period of 2024. This non-cash item increase was primarily due to the recognition of an impairment loss on goodwill arising from the acquisition of Wannaby in January 2025.
Operating Loss
Total operating loss was $0.6 million for the three months ended December 31, 2025, compared with an operating loss of $0.5 million in the same period of 2024, representing an increase of $0.1 million. The increase in operating loss was primarily driven by the recognition of an impairment loss of $2.0 million relating to goodwill arising from the acquisition of Wannaby in 2025.
Net Income
Net income was $0.1 million for the three months ended December 31, 2025, compared to $1.1 million during the same period of 2024, a decrease of 94.2%. The decrease in net income was primarily due to the recognition of an impairment loss of $2.0 million relating to goodwill arising from the acquisition of Wannaby in 2025 and the lower interest income resulting from a decline in interest rates.
Operating Cash Flow
Operating cash flow was $2.6 million for the three months ended December 31, 2025, compared to $3.3 million in the same period of 2024, a decrease of 21.6%.
Financial Results for the Year Ended December 31, 2025
Revenue
Total revenue was $69.2 million for the year ended December 31, 2025, compared to $60.2 million in the same period of 2024, an increase of 14.9%.
AI- and AR- cloud solutions and subscription revenue was $61.1 million for the year ended December 31, 2025, compared to $53.8 million in the same period of 2024, an increase of 13.5%. The increase was driven by the continued revenue growth of YouCam mobile apps and web services subscriptions.
Licensing revenue was $5.3 million for the year ended December 31, 2025, compared to $5.2 million in the same period of 2024, an increase of 1.7%.
Others revenue was $2.8 million for the year ended December 31, 2025, compared to $1.2 million in the same period of 2024, an increase of 133.8%. The increase was primarily driven by the growth of virtual points purchased and consumed by end users. Virtual points are used for AI-powered services available on YouCam mobile apps and web services.
Gross Profit
Gross profit was $53.5 million for the year ended December 31, 2025, compared with $46.9 million in the same period of 2024, an increase of 14.0%. Gross margin was 77.4% for the year ended December 31, 2025, a slight decrease compared to 78.0% in the same period of 2024.
Total Operating Expenses
Total operating expenses were $55.3 million for the year ended December 31, 2025, compared with $50.1 million in the same period of 2024, an increase of 10.3%. The increase was primarily due to increases in research and development expenses and sales and marketing expenses, which were partially offset by a decrease in general and administrative expenses during the same period.
Sales and marketing expenses were $30.8 million for the year ended December 31, 2025, compared to $28.2 million during the same period of 2024, an increase of 9.2%.
Research and development expenses were $15.4 million for the year ended December 31, 2025, compared to $12.0 million during the same period of 2024, an increase of 28.4%.
General and administrative expenses were $7.0 million for the year ended December 31, 2025, compared to $8.5 million during the same period of 2024, a decrease of 17.7%.
Impairment loss on goodwill was $2.0 million for the year ended December 31, 2025. No such impairment was recorded in the same period of 2024. This non-cash item increase was driven by the recognition of an impairment loss on goodwill arising from the acquisition of Wannaby in 2025.
Operating Loss
Total operating loss was $1.7 million for the year ended December 31, 2025, compared with an operating loss of $3.1 million in the same period of 2024, a decrease of $1.4 million. The decrease in operating loss was primarily driven by higher revenue and gross profit, with operating expenses growing at a more moderate pace, which was partially offset by recognition of an impairment loss of $2.0 million on goodwill arising from the acquisition of Wannaby in 2025.
Net Income
Net income was $4.6 million for the year ended December 31, 2025, compared to $5.0 million during the same period of 2024, a decrease of 7.5%.
Operating Cash Flow
Operating cash flow was $13.3 million for the year ended December 31, 2025, compared to $13.0 million in the same period of 2024, an increase of 2.3%. The Company continues to invest in growth while maintaining a healthy cash flow to support business operations underscoring the Company’s operational health and sustainability.
Capital Resource
As of December 31, 2025, the Company’s cash and cash equivalents remained stable at $126.0 million (or $172.4 million when including 6-month time deposits of $36.3 million and US Treasuries of $10.2 million, which are classified as current and non-current financial assets at amortized cost under IFRS, respectively), compared to $127.1 million (or $165.9 million when including time deposits and money market funds) as of December 31, 2024.
Key Business Metrics
The number of active subscribers for the Company’s YouCam mobile apps and web services was 908,000 as of December 31, 2025, compared to over 946,000 as of September 30, 2025, a decrease of 4.0%. This decline was a result of the mobile app subscription plan’s average selling price increase initiative introduced in early 2025, which strategically prioritized higher revenue per user and long-term monetization efficiency over short-term volume growth.
As of December 31, 2025, the Company’s cumulative customer base included 859 brand clients, with over 982,000 digital stock keeping units (“SKUs”) for makeup, haircare, skincare, shoes, bags, eyewear, watches and jewelry products, compared to 842 brand clients and over 953,000 digital SKUs as of September 30, 2025. The number of Key Customers 1 of the Company as of December 31, 2025 was 135 compared to 142 as of September 30, 2025. The decline in the number of Key Customers was primarily due to certain customers being downgraded as a result of lower spending during the period.
____________________
1
“Key Customers” refers to the Company’s brand customers who contributed revenue of more than $50,000 in the trailing 12 months ended on the measurement date.
CEO Remarks and Business Outlook for 2026
Ms. Alice H. Chang, Founder, Chairwoman, and Chief Executive Officer of Perfect Corp., commented, “Perfect Corp. closed 2025 on a strong note, exceeding our full-year guidance and demonstrating the strength of our execution. Our results for the year were driven primarily by continued growth in our B2C mobile apps and web service subscriber base, reflecting strong demand from individual beauty enthusiasts and consumers who value personalization, performance, and the ability to create customized content powered by generative AI. Our sustained focus on AI remains a core driver of innovation across the business, and this momentum positions us well as we enter the next phase of growth, with an increased focus on Agentic AI and API-based solutions.
“We continue to invest in the development of new products and services, including Generative AI beauty solutions, while driving greater operational efficiencies across the organization. This disciplined execution delivered strong revenue growth, a meaningful improvement in company operation, reduction of operating loss, and sustained cash flow generation. As a result, we ended 2025 with a strong cash position, providing the flexibility to invest strategically and support our long-term growth objectives. While the Company reported an operating loss for the period, this was primarily driven by an impairment loss of goodwill charge related to the acquisition of Wannaby. Excluding this non-cash item, the Company would have generated operating income for the fourth quarter and full year of 2025. Perfect Corp. will continue to work toward operating income under IFRS reporting standards in the near term, reflecting the scalability and discipline of its business model. Reaching this milestone would mark a pivotal moment in the Company’s journey, validating years of investment in platform development, AI innovation, and go-to-market execution.
“Our B2C app and web subscription business continues to be the primary driver of growth in 2025, with increases in both revenue per user and user engagement following the price adjustment implemented last year. While subscriber churn was modestly higher, we are seeing an increase in demand for AI-driven image and video editing and creation, reflecting a continued shift toward creativity and personalization powered by AI. Building on this momentum, we plan to introduce additional generative AI capabilities—such as more personalized interactions with our AI Agent and expanded video-mode support—further enhancing the functionality and value of our apps. Our YouCam app suite continues to set the standard for AI-powered creativity and self-expression, offering some of the most popular features in the market, including face reshaping, wrinkle removal, image-to-video, text-to-image, and image-to-image editing and creation. Central to these capabilities is YouCam’s AI Agent, powered by third party large language models (LLMs), which enables users to enhance and edit photos, generate videos, or create AI images simply from a text prompt. Together, these tools deliver a seamless, intelligent, and highly personalized experience that empowers users to express themselves in entirely new ways.
“Perfect’s Beauty AI Agent goes beyond a traditional LLM application by adding a purpose-built intelligence layer on top of the core model. Rather than relying solely on prompt engineering and linear processing, we apply context engineering to modularize inputs, classify intent, and route tasks through parallel sub-agents—resulting in faster response times, higher precision, and reduced hallucination risk. By combining Retrieval-Augmented Generation (RAG) with precision engineering, our agent doesn’t just talk; it can see, score, and recommend with proven accuracy across product virtual try-ons and skin analysis use cases. This architecture positions Perfect Corp. as a Beauty AI infrastructure, delivered through enterprise-grade Widgets, Software Development Kits (SDKs), and Application Programming Interfaces (APIs), and designed with brand safety, governance, and auditability at its core.
“As previously mentioned, our API business is steadily taking shape across multiple growth vectors. Since 2025, Perfect Corp. has built a comprehensive API suite supporting beauty, skin, jewelry, fashion, shoes and apparel industries. Firstly, our agency strategy allows us to act as a force multiplier by embedding our API-driven solutions directly into agency workflows, enabling faster deployment and broader reach. Secondly, we are expanding into new verticals, including the medical and dermatology segments, where our Skin AI technology enhances patient engagement through advanced visualization and personalized experiences. We are also extending our visual commerce capabilities beyond beauty, using virtual try-on to elevate product visualization across categories such as jewelry, shoes, watches, hair, and accessories, further diversifying our addressable market and long-term growth potential.
“Looking ahead to 2026, we see a strong outlook for our B2C apps and web service subscription business, while the B2B enterprise segment is expected to remain more cautious, with limited near-term growth. Against this backdrop, we are continuing our evolution from a tactical service provider to a strategic technology partner, focused on delivering durable, long-term value for our customers. What began as virtual try-on capabilities has expanded into a comprehensive visual commerce enablement platform, powered by Generative AI and an API-first architecture that integrates seamlessly into our partners’ ecosystems. At the same time, we are progressing beyond siloed point solutions toward a unified AI agent capable of operating across multiple roles—delivering a more intelligent, scalable, and impactful omni-solution for both consumers and brands.”
Business Outlook for 2026
Driven by continued revenue growth in both YouCam mobile apps and web service subscriptions, along with sustained demand for our enterprise solutions, the Company expects the full year 2026 total revenue to increase by approximately 10% with a range of plus or minus 2% compared to full year 2025. This forecast is based on the Company’s current assessment of the market and operational conditions, and that these factors are subject to change.
About Perfect Corp.
Founded in 2015, Perfect Corp. is a leading AI company offering self-developed AI- and AR- powered solutions dedicated to transforming the world with digital tech innovations that make your virtual world beautiful. On Perfect’s direct consumer business side, Perfect operates a family of YouCam consumer apps and web-editing services for photo, video and camera users, centered on unleashing creativity with AI-driven features for creation, beautification and enhancement. On Perfect’s enterprise business side, Perfect empowers major beauty, skincare, fashion, jewelry, and watch brands and retailers by supplying them with omnichannel shopping experiences through AR product try-ons and AI-powered skin diagnostics. With cutting-edge technologies such as Generative AI, real-time facial and hand 3D AR rendering and cloud solutions, Perfect enables a personalized, enjoyable, and engaging shopping journey and helps brands elevate customer engagement, increase conversion rates, and propel sales growth. Throughout this journey, Perfect maintains its unwavering commitment to environmental sustainability and fulfilling social responsibilities. For more information, visit https://ir.perfectcorp.com/.
Forward-Looking Statements
This communication contains forward-looking statements within the meaning of Section 27A of the U.S. Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the U.S. Securities Exchange Act of 1934, as amended, or the Exchange Act, that are based on beliefs and assumptions and on information currently available to Perfect. In some cases, you can identify forward-looking statements by the following words: “may,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “continue,” “ongoing,” “target,” “seek” or the negative or plural of these words, or other similar expressions that are predictions or indicate future events or prospects, although not all forward-looking statements contain these words. Any statements that refer to expectations, projections or other characterizations of future events or circumstances, including strategies or plans, are also forward-looking statements. These statements involve risks, uncertainties and other factors that may cause actual results, levels of activity, performance or achievements to be materially different from those expressed or implied by these forward-looking statements. These statements are based on Perfect’s reasonable expectations and beliefs concerning future events and involve risks and uncertainties that may cause actual results to differ materially from current expectations. These factors are difficult to predict accurately and may be beyond Perfect’s control. Forward-looking statements in this communication or elsewhere speak only as of the date made. New uncertainties and risks arise from time to time, and it is impossible for Perfect to predict these events or how they may affect Perfect. In addition, risks and uncertainties are described in Perfect’s filings with the Securities and Exchange Commission. These filings may identify and address other important risks and uncertainties that could cause actual events and results to differ materially from those contained in the forward-looking statements. Perfect cannot assure you that the forward-looking statements in this communication will prove to be accurate. There may be additional risks that Perfect presently does not know or that Perfect currently does not believe are immaterial that could also cause actual results to differ from those contained in the forward-looking statements. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by Perfect, its directors, officers or employees or any other person that Perfect will achieve its objectives and plans in any specified time frame, or at all. Except as required by applicable law, Perfect does not have any duty to, and does not intend to, update or revise the forward-looking statements in this communication or elsewhere after the date of this communication. You should, therefore, not rely on these forward-looking statements as representing the views of Perfect as of any date subsequent to the date of this communication.
STAFFORD, Texas, Feb. 24, 2026 (GLOBE NEWSWIRE) — Greenwich LifeSciences, Inc. (Nasdaq: GLSI) (the “Company”), a clinical-stage biopharmaceutical company focused on its Phase III clinical trial, FLAMINGO-01, which is evaluating Fast Track designated GLSI-100, an immunotherapy to prevent breast cancer recurrences, today announced that two abstracts have been accepted for presentation at the upcoming American Association for Cancer Research (AACR) Annual Meeting 2026, including two corresponding posters.
The AACR 2026 conference will be held from April 17-22, 2026. The AACR plans to publish the abstract titles on March 17, 2026 at 4:30 pm EST, the abstracts on April 17, 2026 at 3:00 pm EST, and the posters on the date of the presentation at the conference.
CEO Snehal Patel commented, “One of these abstracts will be the first abstract co-authored by the Company and the full Steering Committee of FLAMINGO-01.”
About the AACR Annual Meeting 2026
The AACR is the first and largest cancer research organization dedicated to accelerating the conquest of cancer and has more than 61,000 members residing in 143 countries and territories. The AACR Annual Meeting is the focal point of the cancer research community, where scientists, clinicians, other health care professionals, survivors, patients, and advocates gather to share the latest advances in cancer science and medicine. From population science and prevention; to cancer biology, translational, and clinical studies; to survivorship and advocacy; the AACR Annual Meeting highlights the work of the best minds in cancer research from institutions all over the world.
About FLAMINGO-01 Open Label Phase III Data
More than 1,000 patients have been screened with a current screen rate of approximately 600 patients per year. The 250 patient non-HLA-A*02 arm is now fully enrolled, where all patients received GLSI-100, which is 5 times more treated patients and recurrence rate data than the approximately 50 patients treated in the Phase IIb trial. The Primary Immunization Series (PIS), which includes the first 6 GLSI-100 injections over the first 6 months and is required to reach peak protection, is followed by 5 booster injections given every 6 months to prolong the immune response, thereby providing longer-term protection.
In the non-HLA-A*02 arm, a preliminary analysis of recurrence rates after the PIS is completed shows an approximately 80% reduction in recurrence rate.
This observation is trending similarly to the Phase IIb trial results and hazard ratio where HLA-A*02 patients were treated and where breast cancer recurrences were reduced up to 80% compared to a 20-50% reduction in recurrence rate by other approved products.
The immune response at baseline prior to any GLSI-100 treatment, the increasing immune response during the PIS, and the safety profile of non-HLA-A*02 patients is trending similarly to the HLA-A*02 arms of FLAMINGO-01 and to the Phase IIb study.
Analysis of the open label data from FLAMINGO-01 has been conducted in a manner that maintains the study blind. The open label recurrence rate, immune response, and safety data is based on the patients enrolled to date in FLAMINGO-01 and the data provided by the clinical sites so far, which is not completed or fully reviewed, and is thus preliminary. While comparing any preliminary FLAMINGO-01 data to the Phase IIb clinical trial data may be possible, these preliminary results are not a prediction of future results, and the results at the end of the study may differ.
About GLSI-100 Phase IIb Study
In the prospective, randomized, single-blinded, placebo-controlled, multi-center (16 sites led by MD Anderson Cancer Center) Phase IIb clinical trial of HLA-A*02 breast cancer patients, 46 HER2/neu 3+ over-expressor patients were treated with GLSI-100, and 50 placebo patients were treated with GM-CSF alone. After 5 years of follow-up, there was an 80% or greater reduction in cancer recurrences in the HER2/neu 3+ patients who were treated with GLSI-100, followed, and remained disease free over the first 6 months, which we believe is the time required to reach peak immunity and thus maximum efficacy and protection. The Phase IIb results can be summarized as follows:
80% or greater reduction in metastatic breast cancer recurrence rate over 5 years of follow-up with a peak immune response at 6 months and well-tolerated safety profile.
The PIS elicited a potent immune response as measured by local skin tests and immunological assays.
About FLAMINGO-01 and GLSI-100
FLAMINGO-01 (NCT05232916) is a Phase III clinical trial designed to evaluate the safety and efficacy of Fast Track designated GLSI-100 (GP2 + GM-CSF) in HER2 positive breast cancer patients who had residual disease or high-risk pathologic complete response at surgery and who have completed both neoadjuvant and postoperative adjuvant trastuzumab based treatment. The trial is led by Baylor College of Medicine and currently includes US and European clinical sites from university-based hospitals and academic and cooperative networks with plans to open up to 150 sites globally. In the double-blinded arms of the Phase III trial, approximately 500 HLA-A*02 patients are planned to be randomized to GLSI-100 or placebo, and up to 250 patients of other HLA types are planned to be treated with GLSI-100 in a third arm. The trial has been designed to detect a hazard ratio of 0.3 in invasive breast cancer-free survival, where 28 events will be required. An interim analysis for superiority and futility will be conducted when at least half of those events, 14, have occurred. This sample size provides 80% power if the annual rate of events in placebo-treated subjects is 2.4% or greater.
For more information on FLAMINGO-01, please visit the Company’s website here and clinicaltrials.gov here. Contact information and an interactive map of the majority of participating clinical sites can be viewed under the “Contacts and Locations” section. Please note that the interactive map is not viewable on mobile screens. Related questions and participation interest can be emailed to: flamingo-01@greenwichlifesciences.com
About Breast Cancer and HER2/neu Positivity
One in eight U.S. women will develop invasive breast cancer over her lifetime, with approximately 300,000 new breast cancer patients and 4 million breast cancer survivors. HER2 (human epidermal growth factor receptor 2) protein is a cell surface receptor protein that is expressed in a variety of common cancers, including in 75% of breast cancers at low (1+), intermediate (2+), and high (3+ or over-expressor) levels.
About Greenwich LifeSciences, Inc.
Greenwich LifeSciences is a clinical-stage biopharmaceutical company focused on the development of GP2, an immunotherapy to prevent breast cancer recurrences in patients who have previously undergone surgery. GP2 is a 9 amino acid transmembrane peptide of the HER2 protein, a cell surface receptor protein that is expressed in a variety of common cancers, including expression in 75% of breast cancers at low (1+), intermediate (2+), and high (3+ or over-expressor) levels. Greenwich LifeSciences has commenced a Phase III clinical trial, FLAMINGO-01. For more information on Greenwich LifeSciences, please visit the Company’s website at www.greenwichlifesciences.com and follow the Company’s Twitter at https://twitter.com/GreenwichLS.
Forward-Looking Statement Disclaimer
Statements in this press release contain “forward-looking statements” that are subject to substantial risks and uncertainties. All statements, other than statements of historical fact, contained in this press release are forward-looking statements. Forward-looking statements contained in this press release may be identified by the use of words such as “anticipate,” “believe,” “contemplate,” “could,” “estimate,” “expect,” “intend,” “seek,” “may,” “might,” “plan,” “potential,” “predict,” “project,” “target,” “aim,” “should,” “will,” “would,” or the negative of these words or other similar expressions, although not all forward-looking statements contain these words. Forward-looking statements are based on Greenwich LifeSciences Inc.’s current expectations and are subject to inherent uncertainties, risks and assumptions that are difficult to predict, including statements regarding the intended use of net proceeds from the public offering; consequently, actual results may differ materially from those expressed or implied by such forward-looking statements. Further, certain forward-looking statements are based on assumptions as to future events that may not prove to be accurate. These and other risks and uncertainties are described more fully in the section entitled “Risk Factors” in Greenwich LifeSciences’ Annual Report on the most recent Form 10-K for the year ended December 31, 2024, and other periodic reports filed with the Securities and Exchange Commission. Forward-looking statements contained in this announcement are made as of this date, and Greenwich LifeSciences, Inc. undertakes no duty to update such information except as required under applicable law.
Investor & Public Relations Contact for Greenwich LifeSciences Dave Gentry RedChip Companies Inc. Office: 1-800-RED CHIP (733 2447) Email: dave@redchip.com
CHICAGO, Feb. 23, 2026 (GLOBE NEWSWIRE) — FreightCar America, Inc. (NASDAQ: RAIL), a diversified manufacturer of railroad freight cars, today announced that it will release its fourth quarter and full year 2025 financial results on Monday, March 9, 2026, after the market close, and host a teleconference to discuss its fourth quarter and full year 2025 results on the following day. Teleconference details are as follows:
Please note that the webcast is listen-only and webcast participants will not be able to participate in the question and answer portion of the conference call. Interested parties are asked to dial in approximately 10 to 15 minutes prior to the start time of the call.
An audio replay of the conference call will be available beginning at 3:00 p.m. (Eastern Time) on Tuesday, March 10, 2026, until 11:59 p.m. (Eastern Time) on Tuesday, March 24, 2026. To access the replay, please dial (844) 512-2921 or (412) 317-6671. The replay passcode is 13758379. An archived version of the webcast will also be available on the FreightCar America Investor Relations website.
About FreightCar America
FreightCar America, headquartered in Chicago, Illinois, is a leading designer, producer and supplier of railroad freight cars, railcar parts and components. We also specialize in railcar repairs, complete railcar rebody services and railcar conversions that repurpose idled rail assets back into revenue service. Since 1901, our customers have trusted us to build quality railcars that are critical to economic growth and instrumental to the North American supply chain. To learn more about FreightCar America, visit www.freightcaramerica.com.
Record full year revenue of $888.3 million Full year net income of $73.5 million (Adjusted net income of $81.6 million) Record full year Adjusted EBITDA of $171.3 million
HOUSTON, Feb. 23, 2026 (GLOBE NEWSWIRE) — Great Lakes Dredge & Dock Corporation (“Great Lakes” or the “Company”) (Nasdaq: GLDD), the largest provider of dredging services in the United States, today reported financial results for the fourth quarter and year ended December 31, 2025 and the signing of two international offshore energy contracts.
Fourth Quarter 2025 Highlights
Revenue was $256.5 million
Total operating income was $32.6 million
Net income was $12.6 million
Adjusted net income was $20.7 million
Adjusted EBITDA was $44.0 million
Full Year 2025 Highlights
Revenue was $888.3 million
Total operating income was $127.8 million
Net income was $73.5 million
Adjusted net income was $81.6 million
Adjusted EBITDA was $171.3 million
Backlog as of December 31, 2025 was $888.1 million
Previously Announced Saltchuk Transaction
On February 11, 2026, Great Lakes announced that it had entered into a definitive agreement for Saltchuk Resources, Inc. (“Saltchuk”) to acquire the Company. The closing of the transaction will be subject to customary closing conditions, including the expiration of the Hart-Scott-Rodino Act waiting period and the tender of shares representing at least one share more than a majority of Great Lakes’ outstanding shares of common stock, and is expected to close in Q2 2026.
Offshore Energy
Great Lakes entered into two new international offshore energy contracts with a major offshore wind developer. The awarded work will keep the Acadia utilized in Europe for the majority of 2027.
Operational Update
Fourth Quarter 2025
Revenue was $256.5 million, an increase of $53.7 million from the fourth quarter of 2024. The higher revenue in the fourth quarter of 2025 was due primarily to the first full quarter of the Amelia Island working and higher capital and offshore energy revenue as compared to the same period in the fourth quarter last year, partially offset by lower coastal protection and maintenance project revenue.
Gross profit of $53.6 million increased from the fourth quarter of 2024 gross profit of $48.9 million primarily due to improved project performance. Gross profit margin decreased to 20.9% from 24.1% in the fourth quarter of 2024 primarily due to increased drydocking expenses.
Operating income was $32.6 million, increasing from $30.0 million in the prior year’s fourth quarter primarily due to the improved gross profit partially offset by higher general and administrative expenses primarily due to increased incentive compensation and expenses related to the Saltchuk transaction.
Net income was $12.6 million, decreasing from net income of $19.7 million in the prior year fourth quarter. The decrease was primarily driven by an $8.1 million one-time expense, net of tax impact, from the extinguishment of our second lien notes. Excluding this one-time expense, adjusted net income was $20.7 million.
Full Year 2025
Revenue was $888.3 million, an increase of $125.6 million from 2024. The higher revenue in 2025 was due primarily to the delivery of the Amelia Island and higher capital, coastal protection, and offshore energy revenues, partially offset by decrease in maintenance revenue.
Gross profit and gross profit margin of $203.5 million and 22.9%, respectively, increased from full year 2024 gross profit and gross profit margin of $160.6 million and 21.1%, respectively. The increases were driven by higher revenues as well as improved utilization and project performance in the current year.
Operating income for the full year 2025 was $127.8 million, which is a $35.0 million improvement from the prior year. The year-over-year increase is primarily a result of higher gross profit in the current year when compared to prior year, partially offset by higher general and administrative expenses primarily due to increased incentive pay in the current year when compared to prior year.
Net income for the full year 2025 was $73.5 million, which is a $16.2 million improvement compared to $57.3 million for the full year 2024. This increase is primarily driven by improvements to operating income in the current year when compared to prior year, partially offset by an increase in the income tax provision in the current year when compared to prior year as well as by the $8.1 million one-time expense, net of tax impact, from the extinguishment of our second lien notes. Excluding this one-time expense, adjusted net income was $81.6 million.
Balance Sheet, Backlog & Capital Expenditures
At December 31, 2025, the Company had $13.4 million in cash and cash equivalents, total long-term debt of $378.2 million and liquidity of over $330 million.
At December 31, 2025, the Company had $763.2 million in dredging backlog compared to $1.2 billion at December 31, 2024. Dredging backlog as of December 31, 2025 does not include approximately $200.2 million of awards and options pending.
At December 31, 2025, the Company had $124.8 million in offshore energy backlog compared to $44.9 million at December 31, 2024. Offshore energy backlog does not include approximately $2.9 million of options pending.
Total capital expenditures for the full year 2025 were $143.9 million including $69.1 million for the construction of the Acadia, $32.3 million for the Amelia Island, $13.7 million for support equipment, and $28.8 million for maintenance and growth.
Conference Call Information
As previously communicated, in light of the pending transaction with Saltchuk, Great Lakes will not host a conference call and webcast in conjunction with the quarter and year-end report.
Use of Non-GAAP Measures
Adjusted EBITDA, as provided herein, represents net income from continuing operations, adjusted for net interest expense, income taxes, depreciation and amortization expense, debt extinguishments, accelerated maintenance expense for new international deployments, goodwill or asset impairments and gains on bargain purchase acquisitions. Adjusted EBITDA is not a measure derived in accordance with GAAP. The Company presents Adjusted EBITDA as an additional measure by which to evaluate the Company’s operating trends. The Company believes that Adjusted EBITDA is a measure frequently used to evaluate the performance of companies with substantial leverage and that the Company’s primary stakeholders (i.e., its stockholders, bondholders and banks) use Adjusted EBITDA to evaluate the Company’s period to period performance. Additionally, management believes that Adjusted EBITDA provides a transparent measure of the Company’s recurring operating performance and allows management to readily view operating trends, perform analytical comparisons and identify strategies to improve operating performance. For this reason, the Company uses a measure based upon Adjusted EBITDA to assess performance for purposes of determining compensation under the Company’s incentive plan. Adjusted EBITDA should not be considered an alternative to, or more meaningful than, amounts determined in accordance with GAAP including: (a) net income as an indicator of operating performance or (b) cash flows from operations as a measure of liquidity. As such, the Company’s use of Adjusted EBITDA, instead of a GAAP measure, has limitations as an analytical tool, including the inability to determine profitability or liquidity due to the exclusion of accelerated maintenance expense for new international deployments, goodwill or asset impairments, gains on bargain purchase acquisitions, net interest expense and income tax expense and the associated significant cash requirements and the exclusion of depreciation and amortization, which represent significant and unavoidable operating costs given the level of indebtedness and capital expenditures needed to maintain the Company’s business. For these reasons, the Company uses net income to measure the Company’s operating performance and uses Adjusted EBITDA only as a supplement. Adjusted EBITDA is reconciled to net income in the table of financial results. For further explanation, please refer to the Company’s Securities and Exchange Commission (“SEC”) filings.
Adjusted net income and adjusted diluted earnings per share, as provided herein, represent net income and diluted earnings per share, adjusted for loss on extinguishment of debt. We believe adjusted net income and adjusted diluted earnings per share provide useful information about the Company’s operating performance. We believe excluding the [non-recurring loss on extinguishment of debt] provides investors with an alternative and supplemental understanding of our core business. Adjusted net income and adjusted diluted earnings per share are reconciled to net income and diluted earnings per share in the table of financial results. For further explanation, please refer to the Company’s SEC filings.
The Company
Great Lakes Dredge & Dock Corporation is the largest provider of dredging services in the United States, which is complemented with a long history of performing significant international projects. In addition, Great Lakes is fully engaged in expanding its core business into the offshore energy industry. The Company employs experienced civil, ocean and mechanical engineering staff in its estimating, production and project management functions. In its over 136-year history, the Company has never failed to complete a marine project. Great Lakes owns and operates the largest and most diverse fleet in the U.S. dredging industry, comprised of approximately 200 specialized vessels. Great Lakes has a disciplined training program for engineers that ensures experienced-based performance as they advance through Company operations. The Company’s Incident-and Injury-Free® (IIF®) safety management program is integrated into all aspects of the Company’s culture. The Company’s commitment to the IIF® culture promotes a work environment where employee safety is paramount.
Certain statements in this press release may constitute “forward-looking” statements, as defined in Section 21E of the Securities Exchange Act of 1934 (the “Exchange Act”), the Private Securities Litigation Reform Act of 1995 (the “PSLRA”) or in releases made by SEC, all as may be amended from time to time. Such forward-looking statements involve known and unknown risks, uncertainties and other important factors that could cause the actual results, performance or achievements of Great Lakes and its subsidiaries, or industry results, to differ materially from any future results, performance or achievements expressed or implied by such forward-looking statements. Statements that are not historical fact are forward-looking statements. Forward-looking statements can be identified by, among other things, the use of forward-looking language, such as the words “plan,” “believe,” “expect,” “anticipate,” “intend,” “estimate,” “project,” “may,” “would,” “could,” “should,” “seeks,” “are optimistic,” “commitment to” or “scheduled to,” or other similar words, or the negative of these terms or other variations are being made pursuant to the Exchange Act and the PSLRA with the intention of obtaining of these terms or comparable language, or by discussion of strategy or intentions. These cautionary statements have the benefit of the “safe harbor” provisions of such laws. Great Lakes cautions investors that any forward-looking statements made by Great Lakes are not guarantees or indicative of future performance. Important assumptions and other important factors that could cause actual results to differ materially from those forward-looking statements with respect to Great Lakes include, but are not limited to: failure to satisfy the conditions to the transaction with Saltchuk; uncertainties associated with the transaction with Saltchuk; failure to complete the transaction with Saltchuk within the expected timeframe or at all; provisions in the definitive agreement with Saltchuk limiting our ability to pursue alternatives to the transaction with Saltchuk; restrictions on the conduct of our business under the definitive agreement with Saltchuk; potential lawsuits arising out of the proposed transaction with Saltchuk; stockholders’ inability to benefit from future Company growth if the transaction with Saltchuk is completed; unexpected difficulties related to the transaction with Saltchuk and integration of both companies; a reduction in government funding for dredging and other contracts, or government cancellation of such contracts, or the inability of the Corps to let bids to market; our ability to qualify as an eligible bidder under government contract criteria and to compete successfully against other qualified bidders in order to obtain government dredging and other contracts; the political environment and governmental fiscal and monetary policies; cost over-runs, operating cost inflation and potential claims for liquidated damages, particularly with respect to our fixed price contracts; the timing of our performance on contracts and new contracts being awarded to us; significant liabilities that could be imposed were we to fail to comply with government contracting regulations; project delays related to the increasingly negative impacts of climate change or other unusual, non-historical weather patterns; costs necessary to operate and maintain our existing vessels and the construction of new vessels, including with respect to changes in applicable regulations or standards; equipment or mechanical failures; pandemic, epidemic or outbreak of an infectious disease; disruptions to our supply chain for procurement of new vessel build materials or maintenance on our existing vessels; capital and operational costs due to environmental regulations; market and regulatory responses to climate change, including proposed regulations concerning emissions reporting and future emissions reduction goals; contract penalties for any projects that are completed late; force majeure events, including natural disasters, war and terrorists’ actions; changes in the amount of our estimated backlog; significant negative changes attributable to large, single customer contracts; Our ability to obtain financing for the construction of new vessels; our ability to secure contracts to utilize our new offshore energy vessel; unforeseen delays and cost overruns related to the construction of our new vessels; any failure to comply with the Jones Act provisions on coastwise trade, or if those provisions were modified, repealed or interpreted differently; our ability to comply with anti-discrimination laws, including those pertaining to diversity, equity and inclusion programs; fluctuations in fuel prices, particularly given our dependence on petroleum-based products; impacts of nationwide inflation on procurement of new build and vessel maintenance materials; our ability to obtain bonding or letters of credit and risks associated with draws by the surety on outstanding bonds or calls by the beneficiary on outstanding letters of credit; acquisition integration and consolidation, including transaction expenses, unexpected liabilities and operational challenges and risks; divestitures and discontinued operations, including retained liabilities from businesses that we sell or discontinue; potential penalties and reputational damage as a result of legal and regulatory proceedings; any liabilities imposed on us for the obligations of joint ventures, and similar arrangements and subcontractors; increased costs of certain material used in our operations due to newly imposed tariffs; unionized labor force work stoppages; any liabilities for job-related claims under federal law, which does not provide for the liability limitations typically present under state law; operational hazards, including any liabilities or losses relating to personal or property damage resulting from our operations; our substantial amount of indebtedness, which makes us more vulnerable to adverse economic and competitive conditions; restrictions on the operation of our business imposed by financing terms and covenants; impacts of adverse capital and credit market conditions on our ability to meet liquidity needs and access capital; limitations on our hedging strategy imposed by statutory and regulatory requirements for derivative transactions; foreign exchange risks, in particular, related to the new offshore energy vessel build; losses attributable to our investments in privately financed projects; restrictions on foreign ownership of our common stock; restrictions imposed by Delaware law and our charter on takeover transactions that stockholders may consider to be favorable; restrictions on our ability to declare dividends imposed by our financing agreements or Delaware law; significant fluctuations in the market price of our common stock, which may make it difficult for holders to resell our common stock when they want or at prices that they find attractive; changes in previously recorded net revenue and profit as a result of the significant estimates made in connection with our methods of accounting for recognized revenue; maintaining an adequate level of insurance coverage; our ability to find, attract and retain key personnel and skilled labor; disruptions, failures, data corruption, cyber-based attacks, security breaches, or regulatory non-compliance affecting our information technology and operational technology systems; and failure of our share repurchase program to be fully implemented or enhance long-term shareholder value. For additional information on these and other risks and uncertainties, please see Item 1A. “Risk Factors” of Great Lakes’ Annual Report on our most recent Form 10-K, Item 1A. “Risk Factors” of Great Lakes’ Quarterly Report on Form 10-Q’s and in other securities filings by Great Lakes with the SEC.
Although Great Lakes believes that its plans, intentions and expectations reflected in or suggested by such forward-looking statements are reasonable, actual results could differ materially from a projection or assumption in any forward-looking statements. Great Lakes’ future financial condition and results of operations, as well as any forward-looking statements, are subject to change and inherent risks and uncertainties. The forward-looking statements contained in this press release are made only as of the date hereof and Great Lakes does not have or undertake any obligation to update or revise any forward-looking statements whether as a result of new information, subsequent events or otherwise, unless otherwise required by law.
Additional Information and Where to Find It
The tender offer for the outstanding common stock of the Company has not yet commenced. The communication referenced above are for information purposes only and do not constitute an offer to buy or the solicitation of an offer to sell any securities. The solicitation and the offer to buy shares of Company common stock will be made only pursuant to an offer to purchase and related materials that Saltchuk and its subsidiary (“Sub”) intend to file with the U.S. Securities and Exchange Commission (the “SEC”). If the tender offer is commenced, Saltchuk and Sub will file a Tender Offer Statement on Schedule TO with the SEC, and thereafter the Company will file a Solicitation/Recommendation Statement on Schedule 14D-9 with respect to the tender offer. BEFORE MAKING ANY INVESTMENT DECISION, INVESTORS AND SECURITY HOLDERS OF THE COMPANY ARE URGED TO READ THE TENDER OFFER MATERIALS (INCLUDING AN OFFER TO PURCHASE, A RELATED LETTER OF TRANSMITTAL AND CERTAIN OTHER TENDER OFFER DOCUMENTS), THE SOLICITATION/RECOMMENDATION STATEMENT AND ANY OTHER RELEVANT DOCUMENTS FILED OR TO BE FILED WITH THE SEC CAREFULLY AND IN THEIR ENTIRETY WHEN THEY BECOME AVAILABLE BECAUSE THEY WILL CONTAIN IMPORTANT INFORMATION WHICH SHOULD BE CONSIDERED BEFORE ANY DECISION IS MADE WITH RESPECT TO THE TENDER OFFER. These materials will be sent free of charge to Company stockholders when available, and may also be obtained free of charge by contacting the Company’s Investor Relations Department c/o Eric Birge, Vice President of Investor Relations, Great Lakes Dredge & Dock Corporation, 9811 Katy Freeway, Suite 1200, Houston, Texas, 77024, 313-220-3053 or EMBirge@gldd.com. In addition, all of these materials (and all other tender offer documents filed with the SEC) will be available at no charge from the SEC through its website at www.sec.gov. Copies of the documents filed with the SEC by the Company will also be available free of charge under the “Investors—Financials & Filings—SEC filings” section of the Company’s website at gldd.com.
For further information contact: Eric Birge Vice President of Investor Relations 313-220-3053














")