OVERLAND PARK, Kan.–(BUSINESS WIRE)– SelectQuote, Inc. (NYSE: SLQT) today announced that SelectRx, the company’s full-service medication management pharmacy that serves all 50 states, will open a third fulfillment facility in Olathe, Kansas, to serve its rapidly expanding customer base, alongside its existing facilities in Monaca, Pennsylvania, and Indianapolis, Indiana.
SelectQuote launched the SelectRx business in 2021 as part of its Healthcare Services Division after acquiring two smaller pharmacy operations. In just over three years, SelectRx has grown its membership from under 5,000 to over 75,000 members who receive regular prescription shipments in adherence-friendly packaging. The new 54,000 square foot state-of-the-art facility at 404 W. Frontier Lane in Olathe, Kansas, is expected to open during the first half of calendar 2025 and should significantly expand the pharmacy’s capacity.
SelectQuote plans to add over 50 new positions at the location and is seeking experienced pharmacists, pharmacy fulfillment technicians (to assist in filling, processing, and shipping prescriptions), and various leadership and support roles such as shipping and inventory management, facilities management and information technology.
In 2023, SelectRx was recognized as a Patient-Centered Pharmacy Home (PCPH) by The Compliance Team. PCPH is based on TCT’s CMS-recognized Patient-Centered Medical Home program, which puts special emphasis on health maintenance, preventative screening, medication management, multi-specialty medical services, patient experience reporting, and benchmarking for chronically-ill patients.
SelectQuote CEO Tim Danker commented, “We are thrilled with the rapid growth of our SelectRx business over the past three years. With this new facility, we will be able to extend this highly convenient and adherence-friendly pharmacy solution to many more Seniors who can benefit from it. We are also thrilled to build out this beautiful new facility in our home market of Kansas City.”
About SelectQuote:
Founded in 1985, SelectQuote (NYSE: SLQT) provides solutions that help consumers protect their most valuable assets: their families, health, and property. The company pioneered the model of providing unbiased comparisons from multiple, highly-rated insurance companies allowing consumers to choose the policy and terms that best meet their unique needs. Two foundational pillars underpin SelectQuote’s success: a strong force of highly-trained and skilled agents who provide a consultative needs analysis for every consumer, and proprietary technology that sources and routes high-quality leads.
With an ecosystem offering high touchpoints for consumers across Insurance, Medicare, Pharmacy, and Value-Based Care, the company now has four core business lines: SelectQuote Senior, SelectQuote Healthcare Services, SelectQuote Life, and SelectQuote Auto and Home. SelectQuote Senior serves the needs of a demographic that sees around 10,000 people turn 65 each day with a range of Medicare Advantage and Medicare Supplement plans. SelectQuote Healthcare Services is comprised of the SelectRx Pharmacy, a specialized medication management pharmacy, and Healthcare Select which proactively connects its members with best-in-class healthcare services that fit each member’s unique healthcare needs. The platform improves health outcomes and lowers healthcare costs through proactive engagement and access to high-value healthcare solutions.
Investment further expands industry-leading shopping, enrollment, and engagement platform, delivering a differentiated experience and driving high quality outcomes while ensuring peace of mind in Medicare consumers’ healthcare decisions.
CHICAGO and CLEARWATER, Fla., Sept. 04, 2024 (GLOBE NEWSWIRE) — GoHealth, Inc. (Nasdaq: GOCO), a leading health insurance marketplace, today announced that it entered into a purchase and subscription agreement which will ultimately lead to the acquisition of e-TeleQuote Insurance, Inc., a distinguished name in the Medicare insurance marketplace. The transaction is expected to close on September 30, 2024. This strategic move represents a significant milestone in GoHealth’s mission to deliver unparalleled consumer-centric solutions while reinforcing the companies’ shared values of integrity, empathy, and accountability.
The investment in e-TeleQuote aligns seamlessly with GoHealth’s strategic vision and commitment to enhancing consumer experiences through innovation and operational excellence. Both companies bring to the table a deep reservoir of industry expertise and a complementary set of strengths that will drive mutual growth and deliver even greater value to consumers, especially as the upcoming benefit season expects to bring significant disruption and high demand for a high-quality shopping experience for Medicare consumers.
Complementary Attributes for Enhanced Value GoHealth and e-TeleQuote share a dedication to putting consumers at the center of their operations. “GoHealth’s scale, proprietary technology and operational excellence combined with e-TeleQuote’s established talent and high-quality track record will create a mutually accretive relationship poised to drive better outcomes for and meet the evolving needs of our Medicare consumers,” said Vijay Kotte, CEO of GoHealth.
Consumer-First Orientation “At the heart of this transaction is a shared commitment to a consumer-first approach. Both GoHealth and e-TeleQuote have consistently demonstrated a deep understanding of and responsiveness to consumer needs. We are excited to tap into the power of the proprietary technology at GoHealth, including the PlanFit Checkup, which will better serve our consumers while driving increased efficiency and effectiveness in choosing the Medicare plan that best meets their needs. By combining our resources and expertise, we are poised to collectively elevate our consumer experience and deliver even better tailored solutions that prioritize the well-being of our consumers,” said Craig Uchytil, CEO of e-TeleQuote.
Looking Ahead “As we embark on this exciting new chapter, we remain committed to a seamless integration process that prioritizes the interests of our customers, team members, and stakeholders. We are confident that this acquisition will not only strengthen our market position but also enhance our ability to deliver exceptional value and service not only in this upcoming Annual Enrollment Period but for years to come,” said Kotte.
About GoHealth, Inc. GoHealth is a leading health insurance marketplace and Medicare-focused digital health company. Enrolling in a health insurance plan can be confusing for customers, and the seemingly small differences between plans can lead to significant out-of-pocket costs or lack of access to critical medicines and even providers. GoHealth combines cutting-edge technology, data science and deep industry expertise to build trusted relationships with consumers and match them with the healthcare policy and carrier that is right for them. Since its inception, GoHealth has enrolled millions of people in Medicare plans and individual and family plans. For more information, visit https://www.gohealth.com.
About e-TeleQuote Insurance, Inc. e-TeleQuote is a Medicare Insurance Marketplace which specializes in helping Medicare Beneficiaries compare Medicare Advantage and Medicare Supplement plans from top carriers and enroll in the plan that’s right for them.
Forward-Looking Statements This release contains forward-looking statements. We intend such forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements other than statements of historical facts contained in this press release may be forward-looking statements. Statements regarding our future results of operations and financial position, business strategy and plans and objectives of management for future operations, including, among others, statements regarding our expected growth, investment plans and business transformation are forward-looking statements.
In some cases, you can identify forward-looking statements by terms, such as “may,” “will,” “should,” “expects,” “plans,” “anticipates,” “could,” “intends,” “targets,” “projects,” “contemplates,” “believes,” “estimates,” “predicts,” “potential” or “continue” or the negative of these terms or other similar expressions. Accordingly, we caution you that any such forward-looking statements are not guarantees of future performance and are subject to risks, assumptions and uncertainties that are difficult to predict. Although we believe that the expectations reflected in these forward-looking statements are reasonable as of the date made, actual results may prove to be materially different from the results expressed or implied by the forward-looking statements.
These forward-looking statements speak only as of the date of this release and are subject to a number of important factors that could cause actual results to differ materially from those in the forward-looking statements, including important factors described in the section titled “Risk Factors” in our 2023 Form 10-K, and the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Quarterly Report on Form 10-Q for the quarter ended June 30, 2024, and in our other filings with the Securities and Exchange Commission. Any forward-looking statement speaks only as of the date on which it is made, and, except as otherwise required by law, we do not undertake any obligation to publicly update or review any forward-looking statement, whether as a result of new information, future developments or otherwise. New factors emerge from time to time, and it is not possible for us to predict which will arise. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
Enrollment Completed Four Months Ahead of Schedule
IRVINE, Calif., Sept. 04, 2024 (GLOBE NEWSWIRE) — Eledon Pharmaceuticals, Inc. (“Eledon”) (NASDAQ: ELDN) today announced that it has successfully completed enrollment for its Phase 2 BESTOW clinical trial, which is designed to assess the safety and efficacy of its investigational immunosuppression therapy tegoprubart for the prevention of organ rejection in patients undergoing kidney transplantation. The trial reached its target enrollment of 120 participants approximately four months earlier than originally planned.
“We are very pleased to achieve this critical milestone ahead of schedule in our BESTOW trial,” said David-Alexandre C. Gros, M.D., Chief Executive Officer of Eledon. “The accelerated pace of enrollment reflects the strong interest among both clinicians and patients in new innovative therapies that have the potential to improve outcomes in kidney transplantation compared to current standard of care immunosuppression regimens. We are proud to be leading the effort to transform the prevention of organ rejection and based on the early completion of enrollment, we now anticipate reporting top-line results for the BESTOW trial in the fourth quarter of 2025.”
BESTOW, a multicenter, two-arm, active comparator clinical study, enrolled 120 participants undergoing kidney transplantation at sites in North America, Europe and Latin America to evaluate the safety, pharmacokinetics, and efficacy of tegoprubart, an anti-CD40 ligand antibody, compared to the calcineurin inhibitor tacrolimus. The study’s primary objective is to assess graft function at 12 months post-transplant, as measured by estimated glomerular filtration rate (eGFR), in participants treated with tegoprubart compared to tacrolimus. Research has shown that better graft function as assessed by eGFR has been associated with improved long-term graft survival following kidney transplantation.
“Completing enrollment in the Phase 2 BESTOW trial is a significant achievement for our team and, more importantly, for the transplant community,” said Steve Perrin, Ph.D., Chief Scientific Officer and President of Eledon. “It is a testament to the strong collaboration with our clinical sites and the enthusiasm within the community for advances in immunosuppression therapy, an area of research that has not seen major therapeutic innovation in decades. We are deeply grateful to the patients, their families, and the clinical teams for their continued support in advancing this important study.”
The BESTOW trial builds upon results from Eledon’s ongoing Phase 1b trial presented at the American Transplant Congress (ATC) in June 2024, and further demonstrates that tegoprubart has the potential to provide kidney transplant recipients with a safe and effective alternative to calcineurin inhibitors, which are often associated with side effects such as hyperglycemia, new onset diabetes, hypertension, or tremors. Eledon plans to continue advancing its tegoprubart clinical program with the goal of offering a new standard of care immunosuppression therapy for organ transplant patients.
Eledon is currently conducting the Phase 2 BESTOW trial (NCT05983770), the Phase 1b trial (NCT05027906), and a long-term safety and efficacy extension study (NCT06126380) to evaluate tegoprubart for the prevention of organ rejection in patients receiving a kidney transplant.
About Eledon Pharmaceuticals and tegoprubart
Eledon Pharmaceuticals, Inc. is a clinical stage biotechnology company that is developing immune-modulating therapies for the management and treatment of life-threatening conditions. The Company’s lead investigational product is tegoprubart, an anti-CD40L antibody with high affinity for the CD40 Ligand, a well-validated biological target that has broad therapeutic potential. The central role of CD40L signaling in both adaptive and innate immune cell activation and function positions it as an attractive target for non-lymphocyte depleting, immunomodulatory therapeutic intervention. The Company is building upon a deep historical knowledge of anti-CD40 Ligand biology to conduct preclinical and clinical studies in kidney allograft transplantation, xenotransplantation, and amyotrophic lateral sclerosis (ALS). Eledon is headquartered in Irvine, California. For more information, please visit the Company’s website at www.eledon.com.
This press release contains forward-looking statements that involve substantial risks and uncertainties. Any statements about the company’s future expectations, plans and prospects, including statements about planned clinical trials, the development of product candidates, expected timing for initiation of future clinical trials, expected timing for receipt of data from clinical trials, expected or future results of tegoprubart trials and its ability to prevent rejection in connection with kidney transplantation, as well as other statements containing the words “believes,” “anticipates,” “plans,” “expects,” “estimates,” “intends,” “predicts,” “projects,” “targets,” “looks forward,” “could,” “may,” and similar expressions, constitute forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements are inherently uncertain and are subject to numerous risks and uncertainties, including: risks relating to the safety and efficacy of our drug candidates; risks relating to clinical development timelines, including interactions with regulators and clinical sites, as well as patient enrollment; and risks relating to costs of clinical trials and the sufficiency of the company’s capital resources to fund planned clinical trials. Actual results may differ materially from those indicated by such forward-looking statements as a result of various factors. These risks and uncertainties, as well as other risks and uncertainties that could cause the company’s actual results to differ significantly from the forward-looking statements contained herein, are discussed in our quarterly 10-Q, annual 10-K, and other filings with the U.S. Securities and Exchange Commission, which can be found at www.sec.gov. Any forward-looking statements contained in this press release speak only as of the date hereof and not of any future date, and the company expressly disclaims any intent to update any forward-looking statements, whether as a result of new information, future events or otherwise.
August 6, 2024 – Vancouver, Canada – Century Lithium Corp. (TSXV:LCE) (OTCQX: CYDVF) (Frankfurt: C1Z) (Century Lithium or the Company) is pleased to report the successful addition of a lithium carbonate stage at the Company’s Lithium Extraction Facility (Pilot Plant) in Amargosa Valley, Nevada, USA, part of the Company’s 100%-owned Angel Island Mine (the Project). Prior to this addition, concentrated lithium solutions from the Pilot Plant were treated by Saltworks Inc. at their facility in Richmond B.C. where samples of battery quality lithium carbonate were produced. During the first days of startup of the lithium carbonate stage, Century’s team at the Pilot Plant successfully treated 200 liters of concentrated lithium solution and produced 20 kg of high-grade lithium carbonate onsite.
“At this point in time, the ability to make lithium carbonate at our Pilot Plant is an important step forward, in line with recommendations from our recently completed Feasibility Study” commented President and CEO Bill Willoughby. “The drop in lithium prices over the last year has taken a toll on the share price of all lithium companies. Despite the downturn, domestic production is still key to the security of supply in the U.S. While it is becoming well known that a vast amount of lithium is contained within the claystone deposits of Nevada, the benefit in unlocking these resources is the ability to produce a battery quality lithium product onsite and thereby reducing or eliminating the need for downstream processing.”
The Company continues work at the Pilot Plant, utilizing the Company’s patent-pending process for chloride leaching combined with Direct Lithium Extraction (DLE) to generate data as the Company works to identify further technological breakthroughs to make the extraction of lithium from clay more economic. Adding the lithium carbonate stage at the Pilot Plant is one of the recommendations made in the Feasibility Study, not only to further demonstrate that battery quality lithium carbonate can be made, but to better understand and minimize the recycle streams from the DLE stage through to final product in the process. With the assistance of engineers from Hargrove Engineers and Constructors, Century’s team configured equipment to run 40-liter batches of concentrated lithium solution though precipitation, washing and drying steps. Final assays on the lithium carbonate are pending.
Qualified Person
Todd Fayram, MMSA-QP and Senior Vice President, Metallurgy of Century Lithium is the qualified person as defined by National Instrument 43-101 and has approved the technical information in this release.
ABOUT CENTURY LITHIUM CORP.
Century Lithium Corp. is an advanced stage lithium company, focused on developing its 100%-owned Angel Island Mine in west-central Nevada, USA. Century Lithium recently completed a Feasibility Study on its Clayton Valley Lithium Project and is currently in the permitting stage, with the goal of becoming a domestic producer of lithium for the growing electric vehicle and battery storage market.
ON BEHALF OF CENTURY LITHIUM CORP. WILLIAM WILLOUGHBY, PhD., PE President & Chief Executive Officer
NEITHER THE TSX VENTURE EXCHANGE NOR ITS REGULATION SERVICES PROVIDER ACCEPTS RESPONSIBILITY FOR THE ADEQUACY OR ACCURACY OF THE CONTENT OF THIS NEWS RELEASE.
This release contains certain forward-looking statements within the meaning of applicable Canadian securities legislation. In certain cases, forward-looking statements can be identified by the use of words such as “plans”, “expects” or “does not anticipate”, or “believes”, or variations of such words and phrases or statements that certain actions, events or results “may”, “could”, “would”, “might” or “will be taken”, “occur” or “be achieved” and similar expressions suggesting future outcomes or statements regarding an outlook.
Forward-looking statements relate to any matters that are not historical facts and statements of our beliefs, intentions and expectations about developments, results and events which will or may occur in the future, without limitation, statements with respect to the potential development and value of the Project and benefits associated therewith, statements with respect to the expected project economics for the Project, such as estimates of life of mine, lithium prices, production and recoveries, capital and operating costs, IRR, NPV and cash flows, any projections outlined in the Feasibility Study in respect of the Project, the permitting status of the Project and the Company’s future development plans.
These and other forward-looking statements and information are subject to various known and unknown risks and uncertainties, many of which are beyond the ability of the Company to control or predict, that may cause their actual results, performance or achievements to be materially different from those expressed or implied thereby, and are developed based on assumptions about such risks, uncertainties and other factors set out herein.These risks include those described under the heading “Risk Factors” in the Company’s most recent annual information form and its other public filings, copies of which can be under the Company’s profile at www.sedarplus.com. The Company expressly disclaims any obligation to update-forward-looking information except as required by applicable law. No forward-looking statement can be guaranteed and actual future results may vary materially. Accordingly, readers are advised not to place reliance on forward-looking statements or information. Furthermore, Mineral Resources that are not Mineral Reserves do not have demonstrated economic viability.
LOS ALTOS, Calif., Sept. 03, 2024 (GLOBE NEWSWIRE) — Unicycive Therapeutics, Inc. (Nasdaq: UNCY), a clinical-stage biotechnology company developing therapies for patients with kidney disease (the “Company” or “Unicycive”), today announced that the Company has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for Oxylanthanum Carbonate (OLC) for the treatment of hyperphosphatemia in patients with chronic kidney disease (CKD) on dialysis. Unicycive is seeking FDA approval of OLC via the 505(b)(2) regulatory pathway.
“With this NDA submission, we are excited to be one step closer to our goal of bringing OLC to patients with chronic kidney disease who are living with hyperphosphatemia,” said Shalabh Gupta, MD, Chief Executive Officer of Unicycive. “We believe our data support a differentiated and best-in-class therapy that will maintain phosphate control while reducing the onerous pill burden patients currently have to manage. Over the last several months, our team has worked diligently to reach this milestone, and we are now preparing to launch OLC, if approved. We are also pleased to report that the FDA granted a waiver for the NDA application Prescription Drug User Fee Act (PDUFA) fees which is a significant savings of approximately $4 million.”
The NDA submission package is based on data from three clinical studies (a Phase 1 study in healthy volunteers, a bioequivalence study in healthy volunteers, and a tolerability study of OLC in CKD patients on dialysis), multiple preclinical studies, and the specifications and practices related to chemistry, manufacturing and controls (CMC).
About Oxylanthanum Carbonate (OLC)
Oxylanthanum carbonate is a next-generation lanthanum-based phosphate binding agent utilizing proprietary nanoparticle technology being developed for the treatment of hyperphosphatemia in patients with chronic kidney disease (CKD) on dialysis. OLC has over forty issued and granted patents globally. Its potential best-in-class profile may have meaningful patient adherence benefits over currently available treatment options as it requires a lower pill burden for patients in terms of number and size of pills per dose that are swallowed instead of chewed. Based on a survey conducted in 2022, Nephrologists stated that the greatest unmet need in the treatment of hyperphosphatemia with phosphate binders is a lower pill burden and better patient compliance.1 The global market opportunity for treating hyperphosphatemia is expected to exceed $2.5 billion, with the United States accounting for more than $1 billion of that total2. Despite the availability of several FDA-cleared medications, 75 percent of U.S. dialysis patients fail to achieve the target phosphorus levels recommended by published medical guidelines.
Fosrenol® is a registered trademark of Shire International Licensing BV. 1Reason Research, LLC 2022 survey. Results here. 2Fortune Business InsightsTM, Hyperphosphatemia Treatment Market, 2021-2028
About Hyperphosphatemia
Hyperphosphatemia is a serious medical condition that occurs in nearly all patients with End Stage Renal Disease (ESRD). If left untreated, hyperphosphatemia leads to secondary hyperparathyroidism (SHPT), which then results in renal osteodystrophy (a condition similar to osteoporosis and associated with significant bone disease, fractures and bone pain); cardiovascular disease with associated hardening of arteries and atherosclerosis (due to deposition of excess calcium-phosphorus complexes in soft tissue). Importantly, hyperphosphatemia is independently associated with increased mortality for patients with chronic kidney disease on dialysis. Based on available clinical data to date, over 80% of patients show signs of cardiovascular calcification by the time they become dependent on dialysis.
Dialysis patients are already at an increased risk for cardiovascular disease (because of underlying diseases such as diabetes and hypertension), and hyperphosphatemia further exacerbates this. Treatment of hyperphosphatemia is aimed at lowering serum phosphate levels via two means: (1) restricting dietary phosphorus intake; and (2) using, on a daily basis, and with each meal, oral phosphate binding drugs that facilitate fecal elimination of dietary phosphate rather than its absorption from the gastrointestinal tract into the bloodstream.
About Unicycive Therapeutics
Unicycive Therapeutics is a biotechnology company developing novel treatments for kidney diseases. Unicycive’s lead drug candidate, oxylanthanum carbonate (OLC), is a novel investigational phosphate binding agent being developed for the treatment of hyperphosphatemia in chronic kidney disease patients on dialysis. UNI-494 is a patent-protected new chemical entity in clinical development for the treatment of conditions related to acute kidney injury. For more information, please visit Unicycive.com and follow us on LinkedIn, X, and YouTube.
Forward-looking statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified using words such as “anticipate,” “believe,” “forecast,” “estimated” and “intend” or other similar terms or expressions that concern Unicycive’s expectations, strategy, plans or intentions. These forward-looking statements are based on Unicycive’s current expectations and actual results could differ materially. There are several factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results; our clinical trials may be suspended or discontinued due to unexpected side effects or other safety risks that could preclude approval of our product candidates; risks related to business interruptions, which could seriously harm our financial condition and increase our costs and expenses; dependence on key personnel; substantial competition; uncertainties of patent protection and litigation; dependence upon third parties; and risks related to failure to obtain FDA clearances or approvals and noncompliance with FDA regulations. Actual results may differ materially from those indicated by such forward-looking statements as a result of various important factors, including: the uncertainties related to market conditions and other factors described more fully in the section entitled ‘Risk Factors’ in Unicycive’s Annual Report on Form 10-K for the year ended December 31, 2023, and other periodic reports filed with the Securities and Exchange Commission. Any forward-looking statements contained in this press release speak only as of the date hereof, and Unicycive specifically disclaims any obligation to update any forward-looking statement, whether as a result of new information, future events or otherwise.
Investigator-initiated Phase 2 trial to evaluate TNX-102 SL’s potential to reduce severity of acute stress reaction (ASR) and frequency of acute stress disorder (ASD) and posttraumatic stress disorder (PTSD) expected to begin third quarter 2024
Currently, no medication approved at or near point-of-care to treat patients suffering from traumatic events and support their long-term health
CHATHAM, N.J., Aug. 29, 2024 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP) (Tonix or the Company), a fully-integrated biopharmaceutical company with marketed products and a pipeline of development candidates, presented clinical data on? acute stress reaction and prevention of PTSD data of TNX-102 SL in two poster presentations and presented preclinical data demonstrating automated high-throughput assay enabling screening for therapeutics to accelerate wound healing in a third poster presentation at the 2024 Military Health System Research Symposium (MHSRS), held August 26-29, 2024, in Kissimmee, Fla. Copies of the Company’s posters, titled:
“Two Clinical Trials of Bedtime Sublingual Cyclobenzaprine (TNX-102 SL) in Military-Related Posttraumatic Stress Disorder (PTSD) Provide Rationale to Study TNX-102 SL in the Aftermath of Trauma to Reduce Acute Stress Disorder (ASD) and Prevent PTSD”;
“Development of the AURORA Platform Trial Network to Test Interventions to Reduce Acute Stress Reaction Symptoms, and Illustration of Use Testing Sublingual Cyclobenzaprine TNX-102 SL”;
“Integrating Automated High-Throughput Scratch Assay and Cell Painting for Comprehensive Analysis of Cell Migration and Wound Healing”, are available under the Scientific Presentations tab of the Tonix website at www.tonixpharma.com.
TNX-102 SL is being evaluated for the reduction in severity of acute stress reaction (ASR) and the frequency of acute stress disorder (ASD) and posttraumatic stress disorder (PTSD) when administered within 24 hours of trauma event. In two double-blind, randomized clinical trials of military-related PTSD, TNX-102 SL showed effects on sleep and PTSD symptoms in two and four weeks of treatment1. Supportive data on the effects of TNX-102 SL on reducing PTSD symptoms suggest early intervention immediately after trauma using TNX-102 SL has the potential to reduce ASR/ASD symptoms which are similar to those of PTSD2,3. TNX-102 SL has been well-tolerated with no recognized liability for tolerance or abuse. Data from these trials support testing of TNX-102 SL within 24 hours of index trauma for effects on acute stress reaction (ASR) symptoms and the incidence of PTSD. In the U.S. Department of Defense-funded Optimizing Acute Stress Reaction Interventions (OASIS) trial conducted by the University of North Carolina under an investigator-initiated investigational new drug (IND) application, 14 days of bedtime TNX-102 SL will be dosed and tested in the immediate aftermath of motor vehicle collision. The study will test the potential for TNX-102 SL to target trauma-related sleep disturbance and its ability to facilitate recovery from ASR and to prevent PTSD. The results may ultimately provide military personnel with a new treatment option that, when administered in the early aftermath of a traumatic event to individuals with ASR symptoms, improves warfighter function.
“In previous trials, TNX-102 SL has been shown to improve sleep quality in PTSD and increased activity on sleep and stress-related symptoms in the first several weeks of treatment after a trauma event”, said Seth Lederman, M.D., Chief Executive Officer of Tonix Pharmaceuticals. “Since sleep disturbance plays a critical role in the development and maintenance of PTSD, sleep improvements may reorient the trajectory of posttraumatic pathology from acute trauma towards early recovery. The OASIS study is driven by the observation that the symptoms of ASR and PTSD are similar and by the hypothesis that TNX-102 SL’s effect on sleep quality may reduce ASR symptoms, potentially providing military personnel, veterans, and civilians with a new treatment option that, when administered in the early aftermath of a traumatic event, improves recovery, job performance, and quality of life.”
The investigator-initiated OASIS trial will examine the safety and efficacy of TNX-102 SL to reduce adverse posttraumatic neuropsychiatric sequelae among patients presenting to the emergency department (ED) after a motor vehicle collision. The trial plans to enroll approximately 180 trauma survivors at ED study sites around the U.S. Participants will be randomized in the ED to receive a two-week course of either TNX-102 SL 5.6 mg or placebo. The first participant for the OASIS trial is expected to enroll in the third quarter of 2024.
The OASIS trial will build upon a foundation of knowledge and infrastructure developed through the UNC-led, $40 million AURORA initiative. AURORA is a major national research initiative to improve the understanding, prevention and recovery of individuals who experience a traumatic event. AURORA is supported by funding from the National Institutes of Health (NIH), leading brain health nonprofit One Mind, private foundations, and partnerships with leading tech companies, such as Mindstrong Health and Verily Life Sciences, the healthcare arm of Alphabet, the parent company of Google.
Acute and chronic stress disorders can affect both civilian and military populations. According to the National Center for PTSD, in the U.S. about 60% of men and 50% of women experience at least one trauma in their lives.4 In the U.S. alone, one-third of ED visits (40-50 million patients per year) involve evaluation after trauma exposures, and in a 2014 study involving 3,157 US veterans, 87% reported exposure to at least one potentially traumatic event during their service.5 Moreover, as many as 500,000 U.S. troops who served in wars between 2001 and 2015 were diagnosed with PTSD.6
The third poster, titled “Integrating Automated High-Throughput Scratch Assay and Cell Painting for Comprehensive Analysis of Cell Migration and Wound Healing”, demonstrated optimization of a highly efficient scratch-wound assay development method. The scratch-wound assay, commonly used to study wound healing, has limitations that the study addresses by introducing an automated miniaturized high-throughput wound healing assay, enabling mass screening and identification of novel therapies for wound-healing. The screening technology was merged with cell-painting to allow discovery of morphological characteristics to identify mechanism of action of drugs for wound healing.
Tonix Pharmaceuticals Holding Corp.* Tonix is a fully-integrated biopharmaceutical company focused on developing, licensing and commercializing therapeutics to treat and prevent human disease and alleviate suffering. Tonix recently announced the U.S. Department of Defense (DoD), Defense Threat Reduction Agency (DTRA) awarded it a contract for up to $34 million over five years in an Other Transaction Agreement (OTA) to develop TNX-4200 small molecule broad-spectrum antiviral agents targeting CD45 for the prevention or treatment of infections to improve the medical readiness of military personnel in biological threat environments. Tonix owns and operates a state-of-the art infectious disease research facility in Frederick, MD. The company’s Good Manufacutring Practice (GMP)-capable advanced manufacturing facility in Dartmouth, MA was purpose-built to manufacture TNX-801 and the GMP suites are ready to be reactivated in case of a national or international emergency. Tonix’s development portfolio is focused on central nervous system (CNS) disorders. Tonix’s priority is to submit a New Drug Application (NDA) to the FDA in the second half of 2024 for TNX-102 SL, a product candidate for which two statistically significant Phase 3 studies have been completed for the management of fibromyalgia. The FDA has granted Fast Track designation to TNX-102 SL for the management of fibromyalgia. TNX-102 SL is also being developed to treat acute stress reaction. Tonix’s CNS portfolio includes TNX-1300 (cocaine esterase), a biologic designed to treat cocaine intoxication that has Breakthrough Therapy designation. Tonix’s immunology development portfolio consists of biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases. Tonix also has product candidates in development in the areas of rare disease and infectious disease. Tonix Medicines, our commercial subsidiary, markets Zembrace® SymTouch® (sumatriptan injection) 3 mg and Tosymra® (sumatriptan nasal spray) 10 mg for the treatment of acute migraine with or without aura in adults.
*Tonix’s product development candidates are investigational new drugs or biologics and have not been approved for any indication.
Zembrace SymTouch and Tosymra are registered trademarks of Tonix Medicines. All other marks are property of their respective owners.
This press release and further information about Tonix can be found at www.tonixpharma.com.
TNX-102 SL (cyclobenzaprine HCl sublingual tablets) has not been approved for any indication; (Tonmya™ is conditionally approved by FDA for the management of fibromyalgia)
Sullivan GM, et al. Randomized clinical trial of bedtime sublingual cyclobenzaprine (TNX-102 SL) in military-related PTSD and the role of sleep quality in treatment response. Psychiatry Res. 2021 Jul;301:113974.
Parmenter ME, et al. A phase 3, randomized, placebo-controlled, trial to evaluate the efficacy and safety of bedtime sublingual cyclobenzaprine (TNX-102 SL) in military-related posttraumatic stress disorder. Psychiatry Res. 2024 (In Press). https://doi.org/10.1016/j.psychres.2024.115764
Goldstein RB, et al. Soc Psychiatry Psychiatr Epidemiol. 2016. 51(8):1137-48
Wisco BE, et al. J Clin Psychiatry. 2014. 75(12):1338-46
Thompson M. Time. 2015;185(12):40-3
Forward Looking Statements Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; risks related to the failure to successfully market any of our products; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2023, as filed with the Securities and Exchange Commission (the “SEC”) on April 1, 2024, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
JERICHO, N.Y.–(BUSINESS WIRE)– 1-800-FLOWERS.COM, Inc. (NASDAQ: FLWS), a leading provider of gifts designed to help inspire customers to give more, connect more, and build more and better relationships, today announced that Bill Shea has confirmed his intention to retire as Chief Financial Officer, effective December 29, 2024. The Board of Directors has appointed James Langrock as Chief Financial Officer, effective December 29, 2024.
“I want to take this opportunity to congratulate Bill on his upcoming retirement and to thank him for his three decades of tireless commitment to our company,” said Jim McCann, Chairman and Chief Executive Officer of 1-800-FLOWERS.COM, Inc. “Bill has been a terrific partner to me and a tremendous asset to our company during a period of tremendous growth and transformation. We wish Bill all the best upon his retirement.”
“I also want to congratulate James on his appointment to Chief Financial Officer upon Bill’s retirement,” Mr. McCann continued. “James has been a great addition to our leadership team since joining the Company and brings a great wealth of financial expertise to our company.”
Mr. Langrock joined 1-800-FLOWERS.COM, Inc. in April as Chief Administrative Officer, bringing a broad range of finance experience to the Company. Prior to joining the Company, Mr. Langrock held the position of Chief Financial Officer at several public and privately held companies, including Charcuterie Artisans, The Hain Celestial Group, and Monster Worldwide, Inc.
About 1-800-FLOWERS.COM, Inc.
1-800-FLOWERS.COM, Inc. is a leading provider of gifts designed to help inspire customers to give more, connect more, and build more and better relationships. The Company’s e-commerce business platform features an all-star family of brands, including: 1-800-Flowers.com®, 1-800-Baskets.com®, Cheryl’s Cookies®, Harry & David®, PersonalizationMall.com®, Shari’s Berries®, FruitBouquets.com®, Things Remembered®, Moose Munch®, The Popcorn Factory®, Wolferman’s Bakery®, Vital Choice®, and Simply Chocolate®. Through the Celebrations Passport® loyalty program, which provides members with free standard shipping and no service charge on eligible products across our portfolio of brands, 1-800-FLOWERS.COM, Inc. strives to deepen relationships with customers. The Company also operates BloomNet®, an international floral and gift industry service provider offering a broad-range of products and services designed to help members grow their businesses profitably; Napco℠, a resource for floral gifts and seasonal décor; DesignPac Gifts, LLC, a manufacturer of gift baskets and towers; and Alice’s Table®, a lifestyle business offering fully digital livestreaming and on demand floral, culinary and other experiences to guests across the country. 1-800-FLOWERS.COM, Inc. was recognized among America’s Most Trustworthy Companies by Newsweek. 1-800-FLOWERS.COM, Inc. was also recognized among the top 5 on the National Retail Federation’s 2021 Hot 25 Retailers list, which ranks the nation’s fastest-growing retail companies, and was named to the Fortune 1000 list in 2022. Shares in 1-800-FLOWERS.COM, Inc. are traded on the NASDAQ Global Select Market, ticker symbol: FLWS. For more information, visit 1800flowersinc.com.
FLWS–COMP FLWS-FN
Special Note Regarding Forward Looking Statements:
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements represent the Company’s current expectations or beliefs concerning future events and can generally be identified using statements that include words such as “estimate,” “expects,” “project,” “believe,” “anticipate,” “intend,” “plan,” “foresee,” “forecast,” “likely,” “should,” “will,” “target” or similar words or phrases. These forward-looking statements are subject to risks, uncertainties, and other factors, many of which are outside of the Company’s control, which could cause actual results to differ materially from the results expressed or implied in the forward-looking statements, including, but not limited to, statements regarding the Company’s ability to achieve its guidance for the full Fiscal year; the Company’s ability to leverage its operating platform and reduce its operating expense ratio; its ability to successfully integrate acquired businesses and assets; its ability to successfully execute its strategic initiatives; its ability to cost effectively acquire and retain customers; the outcome of contingencies, including legal proceedings in the normal course of business; its ability to compete against existing and new competitors; its ability to manage expenses associated with sales and marketing and necessary general and administrative and technology investments; its ability to reduce promotional activities and achieve more efficient marketing programs; and general consumer sentiment and industry and economic conditions that may affect levels of discretionary customer purchases of the Company’s products. The Company undertakes no obligation to publicly update any of the forward-looking statements, whether because of new information, future events or otherwise, made in this release or in any of its SEC filings. Consequently, you should not consider any such list to be a complete set of all potential risks and uncertainties. For a more detailed description of these and other risk factors, refer to the Company’s SEC filings, including the Company’s Annual Reports on Form 10-K and its Quarterly Reports on Form 10-Q.
Reports Fiscal Year 2024 Revenue of $1.83 Billion and a Net Loss of $6.1 Million, which Includes a Non-Cash Impairment Charge of $19.8 million Recorded in the Second Quarter
Fiscal Year 2024 Gross Profit Margin Increased 260 Basis Points to 40.1%
Fiscal Year 2024 Adjusted EBITDA1 Increased to $93.1 million
Issues Fiscal Year 2025 Outlook
(1) Refer to “Definitions of Non-GAAP Financial Measures” and the tables attached at the end of this press release for reconciliation of non-GAAP results to applicable GAAP results.)
JERICHO, N.Y.–(BUSINESS WIRE)– 1-800-FLOWERS.COM, Inc. (NASDAQ: FLWS), a leading provider of gifts designed to help inspire customers to give more, connect more, and build more and better relationships, today reported results for its Fiscal 2024 fourth quarter and year ended June 30, 2024.
“In a dynamic consumer environment that impacted discretionary consumer spending, especially amongst lower income households, our organization was able to grow year-over-year adjusted EBITDA, which benefitted from our significant gross margin recovery and our expense optimization efforts that more than offset the top line decline,” said Jim McCann, Chairman and Chief Executive Officer of 1-800-FLOWERS.COM, Inc. “During Fiscal 2024, through our Relationship Innovation initiatives, we significantly enhanced our gifting platform, including category expansion, broadening our price points, increasing our assortment of gifts available for same-day delivery, and enhancing the user experience. We also experienced a significant recovery in our gross profit margin, which benefitted from a reversion to the mean on a number of commodity costs combined with our Work Smarter initiatives to operate more efficiently.”
“As we turn to Fiscal 2025, with our gross margin recovery well underway, our organization continues to be keenly focused on improving our sales trends by leveraging our Relationship Innovation initiatives. Acknowledging the uncertain consumer environment, we anticipate revenue trends improving as the fiscal year progresses as consumers respond to our newer offerings and services. We are confident in our strategic direction to be the gifting destination of choice for thoughtful and expressive gift-giving occasions and remain focused on delivering long-term value to our shareholders,” Mr. McCann continued.
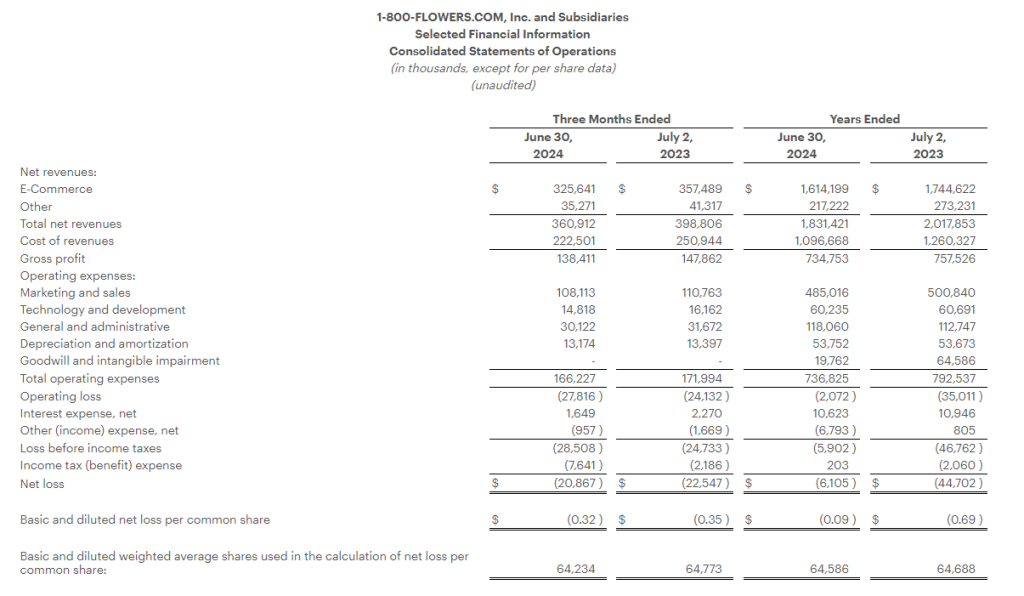
Fiscal 2024 Fourth Quarter Highlights
Total consolidated revenues decreased 9.5% to $360.9 million, compared with total consolidated revenues of $398.8 million in the prior year period.
Gross profit margin increased 130 basis points to 38.4%, compared with 37.1% in the prior year period. The gross profit margin improved on lower freight costs, a decline in certain commodity costs, and the Company’s logistics optimization efforts.
Operating expenses declined $5.8 million to $166.2 million, as compared with the prior year period.
Net loss for the quarter was $20.9 million, or ($0.32) per share, as compared to a net loss of $22.5 million, or ($0.35) per share in the prior year period.
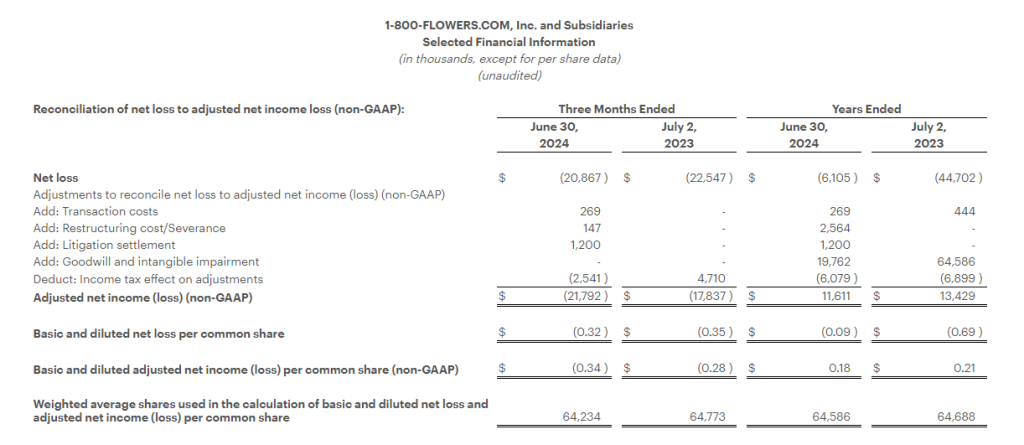
Adjusted Net Loss1 was $21.8 million, or ($0.34) per share, compared with an Adjusted Net Loss1 of $17.8 million, or ($0.28) per share, in the prior year period.
Adjusted EBITDA1 loss for the quarter was $8.8 million, as compared with an Adjusted EBITDA1 loss of $6.6 million in the prior year period.
Acquired Scharffen Berger Chocolate Maker, a high-end producer of extraordinary craft chocolates, that enhances and expands the Company’s chocolate offerings within its gourmet food and gift basket business. The acquisition closed after the fourth quarter ended.
Fiscal Year 2024 Highlights
Total consolidated revenues decreased 9.2% to $1.83 billion, compared with total consolidated revenues of $2.02 billion in the prior year period.
Gross profit margin increased 260 basis points to 40.1%, compared with 37.5% in the prior year period. The gross profit margin improved on lower freight costs, improved commodity costs, and the Company’s logistics optimization efforts.
Operating expenses declined $55.7 million to $736.8 million, as compared with the prior year period. Excluding impairment and other non-recurring charges in both periods, as well as the impact of the Company’s non-qualified deferred compensation plan in both periods, operating expenses declined by $22.2 million to $706.1 million, as compared with the prior year.
Net loss for the fiscal year was $6.1 million, or ($0.09) per share, compared with $44.7 million, or ($0.69) per share, in the prior year period. Both periods include impairment charges as outlined in the financial tables.
Adjusted Net Income1 was $11.6 million, or $0.18 per share, compared with Adjusted Net Income1 of $13.4 million, or $0.21 per share, in the prior year period.
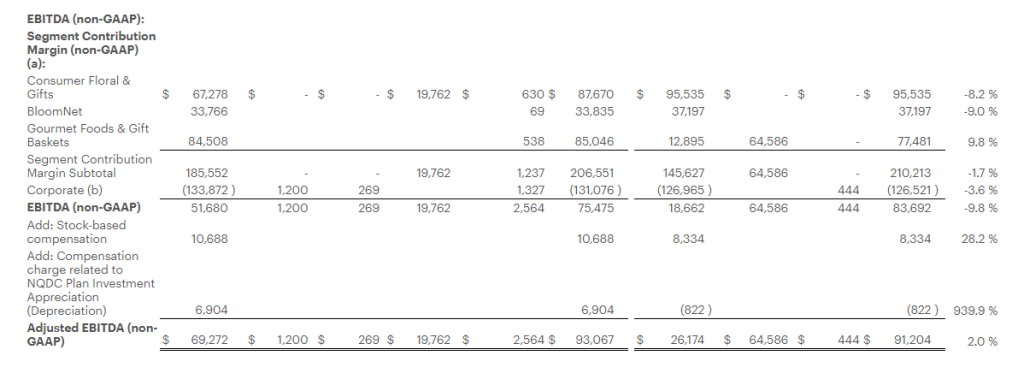
Adjusted EBITDA1 for the fiscal year was $93.1 million, as compared with $91.2 million in the prior year period.
Net cash provided by operating activities was $95.0 million.
Generated Free Cash Flow1 of $56.4 million.
Segment Results
The Company provides Fiscal 2024 fourth quarter and full year selected financial results for its Gourmet Foods and Gift Baskets, Consumer Floral and Gifts, and BloomNet segments in the tables attached to this release and as follows:
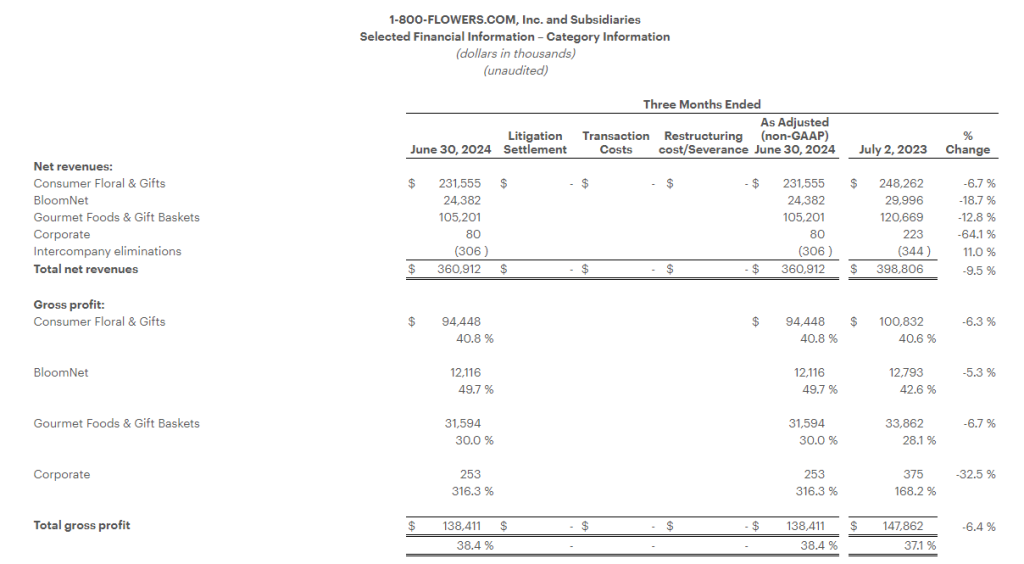
Gourmet Foods and Gift Baskets: Revenues for the quarter declined 12.8% to $105.2 million, as compared with the prior year period. Gross profit margin increased 190 basis points from the prior year period to 30.0%, benefiting from lower freight costs, the Company’s inventory and labor optimization efforts, as well as a decline in certain commodity costs. Segment contribution margin1 loss was $14.4 million, compared with a loss of $13.4 million in the prior year period.
For the full fiscal year, revenue decreased 9.4% to $874.3 million. Gross profit margin increased 340 basis points to 38.3%, benefiting from lower freight costs, the Company’s inventory and labor optimization efforts, as well as a decline in certain commodity costs. Excluding the impact of the severance charge in the current year and the impairment charge a year ago, segment contribution margin1 for the year was $85.0 million, compared with $77.5 million in the prior year.
Consumer Floral & Gifts: Revenues for the quarter declined 6.7% to $231.6 million, as compared with the prior year period. Gross profit margin increased 20 basis points from the prior year period to 40.8%. Segment contribution margin1 was $25.7 million, compared with $30.7 million in the prior year period.
For the full fiscal year, revenues decreased 7.7% to $849.8 million, as compared with the prior year period. Gross profit margin increased 130 basis points from the prior year period to 40.8%, improving on lower fulfillment costs and the Company’s logistics optimization efforts. Excluding the impact of the severance and impairment charges in the current year, segment contribution margin1 was $87.7 million, compared with $95.5 million in the prior year.
BloomNet: Revenues for the quarter declined 18.7% to $24.4 million, as compared with the prior year period. Revenue and gross margin were impacted by the lower volume of lower margin orders processed by BloomNet. Gross profit margin increased 710 basis points from the prior year period to 49.7%, also benefitting from lower ocean freight costs as well as product mix. Segment contribution margin1 was $7.8 million, compared with $7.4 million in the prior year period.
For the year, revenues decreased 19.1% to $107.8 million, as compared with the prior year period. Gross profit margin increased 550 basis points from the prior year period to 48.2% due to lower volume of lower margin orders, lower ocean freight costs, as well as product mix. Excluding the impact of the severance charge in the current year, segment contribution margin1 for the year was $33.8 million, compared with $37.2 million in the prior year.
Company Guidance
For Fiscal 2025, with a sustained challenging consumer environment, the Company expects revenue trends to improve as the fiscal year progresses benefitting from the company’s Relationship Innovation initiatives that have expanded the Company’s product offerings, broadened price points, and enhanced the user experience, combined with increased marketing spend. Additionally, the guidance assumes increased incentive compensation expense.
As a result, for Fiscal Year 2025 the Company expects:
total revenues on a percentage basis to be in a range of flat to a decrease in the low-single digits, as compared with the prior year;
Adjusted EBITDA1 to be in a range of $85 million to $95 million; and
Free Cash Flow1 to be in a range of $45 million to $55 million.
Conference Call
The Company will conduct a conference call to discuss the above details and attached financial results today, August 29, 2024, at 8:00 a.m. (ET). The conference call will be webcast from the Investors section of the Company’s website at www.1800flowersinc.com. A recording of the call will be posted on the Investors section of the Company’s website within two hours of the call’s completion. A telephonic replay of the call can be accessed beginning at 2:00 p.m. (ET) today through September 5, 2024, at: (US) 1-877-344-7529; (Canada) 855-669-9658; (International) 1-412-317-0088; enter conference ID #: 5141252.
Definitions of non-GAAP Financial Measures:
We sometimes use financial measures derived from consolidated financial information, but not presented in our financial statements prepared in accordance with U.S. generally accepted accounting principles (“GAAP”). Certain of these are considered “non-GAAP financial measures” under the U.S. Securities and Exchange Commission rules. Non-GAAP financial measures referred to in this document are either labeled as “non-GAAP” or designated as such with a “1”. See below for definitions and the reasons why we use these non-GAAP financial measures. Where applicable, see the Selected Financial Information below for reconciliations of these non-GAAP measures to their most directly comparable GAAP financial measures. Reconciliations for forward-looking figures would require unreasonable efforts at this time because of the uncertainty and variability of the nature and amount of certain components of various necessary GAAP components, including, for example, those related to compensation, tax items, amortization or others that may arise during the year, and the Company’s management believes such reconciliations would imply a degree of precision that would be confusing or misleading to investors. For the same reasons, the Company is unable to address the probable significance of the unavailable information. The lack of such reconciling information should be considered when assessing the impact of such disclosures.
EBITDA and Adjusted EBITDA:
We define EBITDA as net income (loss) before interest, taxes, depreciation, and amortization. Adjusted EBITDA is defined as EBITDA adjusted for the impact of stock-based compensation, Non-Qualified Plan Investment appreciation/depreciation, and for certain items affecting period-to-period comparability. See Selected Financial Information for details on how EBITDA and Adjusted EBITDA were calculated for each period presented. The Company presents EBITDA and Adjusted EBITDA because it considers such information meaningful supplemental measures of its performance and believes such information is frequently used by the investment community in the evaluation of similarly situated companies. The Company uses EBITDA and Adjusted EBITDA as factors to determine the total amount of incentive compensation available to be awarded to executive officers and other employees. The Company’s credit agreement uses EBITDA and Adjusted EBITDA to determine its interest rate and to measure compliance with certain covenants. EBITDA and Adjusted EBITDA are also used by the Company to evaluate and price potential acquisition candidates. EBITDA and Adjusted EBITDA have limitations as analytical tools and should not be considered in isolation or as a substitute for analysis of the Company’s results as reported under GAAP. Some of the limitations are: (a) EBITDA and Adjusted EBITDA do not reflect changes in, or cash requirements for, the Company’s working capital needs; (b) EBITDA and Adjusted EBITDA do not reflect the significant interest expense, or the cash requirements necessary to service interest or principal payments, on the Company’s debts; and (c) although depreciation and amortization are non-cash charges, the assets being depreciated and amortized may have to be replaced in the future and EBITDA does not reflect any cash requirements for such capital expenditures. EBITDA and Adjusted EBITDA should only be used on a supplemental basis combined with GAAP results when evaluating the Company’s performance.
Segment Contribution Margin and Adjusted Segment Contribution Margin
We define Segment Contribution Margin as earnings before interest, taxes, depreciation, and amortization, before the allocation of corporate overhead expenses. Adjusted Segment Contribution Margin is defined as Segment Contribution Margin adjusted for certain items affecting period-to-period comparability. See Selected Financial Information for details on how Segment Contribution Margin and Adjusted Segment Contribution Margin were calculated for each period presented. When viewed together with our GAAP results, we believe Segment Contribution Margin and Adjusted Segment Contribution Margin provide management and users of the financial statements meaningful information about the performance of our business segments. Segment Contribution Margin and Adjusted Segment Contribution Margin are used in addition to and in conjunction with results presented in accordance with GAAP and should not be relied upon to the exclusion of GAAP financial measures. The material limitation associated with the use of Segment Contribution Margin and Adjusted Segment Contribution Margin is that they are an incomplete measure of profitability as they do not include all operating expenses or non-operating income and expenses. Management compensates for this limitation when using these measures by looking at other GAAP measures, such as Operating Income and Net Income.
Adjusted Net Income (Loss) and Adjusted or Comparable Net Income (Loss) Per Common Share:
We define Adjusted Net Income (Loss) and Adjusted or Comparable Net Income (Loss) Per Common Share as Net Income (Loss) and Net Income (Loss) Per Common Share adjusted for certain items affecting period-to-period comparability. See Selected Financial Information below for details on how Adjusted Net Income (Loss) Per Common Share and Adjusted or Comparable Net Income (Loss) Per Common Share were calculated for each period presented. We believe that Adjusted Net Income (Loss) and Adjusted or Comparable Net Income (Loss) Per Common Share are meaningful measures because they increase the comparability of period-to-period results. Since these are not measures of performance calculated in accordance with GAAP, they should not be considered in isolation of, or as a substitute for, GAAP Net Income (Loss) and Net Income (Loss) Per Common share, as indicators of operating performance and they may not be comparable to similarly titled measures employed by other companies.
Free Cash Flow:
We define Free Cash Flow as net cash provided by operating activities less capital expenditures. The Company considers Free Cash Flow to be a liquidity measure that provides useful information to management and investors about the amount of cash generated by the business after the purchases of fixed assets, which can then be used to, among other things, invest in the Company’s business, make strategic acquisitions, strengthen the balance sheet, and repurchase stock or retire debt. Free Cash Flow is a liquidity measure that is frequently used by the investment community in the evaluation of similarly situated companies. Since Free Cash Flow is not a measure of performance calculated in accordance with GAAP, it should not be considered in isolation or as a substitute for analysis of the Company’s results as reported under GAAP. A limitation of the utility of Free Cash Flow as a measure of financial performance is that it does not represent the total increase or decrease in the Company’s cash balance for the period.
About 1-800-FLOWERS.COM, Inc.
1-800-FLOWERS.COM, Inc. is a leading provider of gifts designed to help inspire customers to give more, connect more, and build more and better relationships. The Company’s e-commerce business platform features an all-star family of brands, including: 1-800-Flowers.com®, 1-800-Baskets.com®, Cheryl’s Cookies®, Harry & David®, PersonalizationMall.com®, Shari’s Berries®, FruitBouquets.com®, Things Remembered®, Moose Munch®, The Popcorn Factory®, Wolferman’s Bakery®, Vital Choice®, and Simply Chocolate®. Through the Celebrations Passport® loyalty program, which provides members with free standard shipping and no service charge on eligible products across our portfolio of brands, 1-800-FLOWERS.COM, Inc. strives to deepen relationships with customers. The Company also operates BloomNet®, an international floral and gift industry service provider offering a broad-range of products and services designed to help members grow their businesses profitably; Napco℠, a resource for floral gifts and seasonal décor; DesignPac Gifts, LLC, a manufacturer of gift baskets and towers; and Alice’s Table®, a lifestyle business offering fully digital livestreaming and on demand floral, culinary and other experiences to guests across the country. 1-800-FLOWERS.COM, Inc. was recognized among America’s Most Trustworthy Companies by Newsweek. 1-800-FLOWERS.COM, Inc. was also recognized among the top 5 on the National Retail Federation’s 2021 Hot 25 Retailers list, which ranks the nation’s fastest-growing retail companies, and was named to the Fortune 1000 list in 2022. Shares in 1-800-FLOWERS.COM, Inc. are traded on the NASDAQ Global Select Market, ticker symbol: FLWS. For more information, visit 1800flowersinc.com or follow @1800FLOWERSInc on Twitter.
FLWS–COMP FLWS-FN
Special Note Regarding Forward Looking Statements:
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements represent the Company’s current expectations or beliefs concerning future events and can generally be identified using statements that include words such as “estimate,” “expects,” “project,” “believe,” “anticipate,” “intend,” “plan,” “foresee,” “forecast,” “likely,” “should,” “will,” “target” or similar words or phrases. These forward-looking statements are subject to risks, uncertainties, and other factors, many of which are outside of the Company’s control, which could cause actual results to differ materially from the results expressed or implied in the forward-looking statements, including, but not limited to, statements regarding the Company’s ability to achieve its guidance for the full Fiscal year; the Company’s ability to leverage its operating platform and reduce its operating expense ratio; its ability to successfully integrate acquired businesses and assets; its ability to successfully execute its strategic initiatives; its ability to cost effectively acquire and retain customers; the outcome of contingencies, including legal proceedings in the normal course of business; its ability to compete against existing and new competitors; its ability to manage expenses associated with sales and marketing and necessary general and administrative and technology investments; its ability to reduce promotional activities and achieve more efficient marketing programs; and general consumer sentiment and industry and economic conditions that may affect levels of discretionary customer purchases of the Company’s products. The Company undertakes no obligation to publicly update any of the forward-looking statements, whether because of new information, future events or otherwise, made in this release or in any of its SEC filings. Consequently, you should not consider any such list to be a complete set of all potential risks and uncertainties. For a more detailed description of these and other risk factors, refer to the Company’s SEC filings, including the Company’s Annual Reports on Form 10-K and its Quarterly Reports on Form 10-Q.
Note: The following tables are an integral part of this press release without which the information presented in this press release should be considered incomplete.
LIMASSOL, Cyprus, Aug. 28, 2024 (GLOBE NEWSWIRE) — GDEV Inc. (NASDAQ: GDEV), an international gaming and entertainment company (“GDEV” or the “Company”), announces that its financial results for the second quarter ended June 30, 2024 will be released at 8:00 a.m. (Eastern Time) on Wednesday, September 4, 2024.
GDEV will host a conference call and webcast to discuss its results at 09:00 a.m. U.S. Eastern Time the same day.
The press release, as well as supplementary slides will be available at gdev.inc. To listen to the audio webcast please follow this link. To participate in the conference call, please use the following details:
US toll-free dial: +1 844-543-0451 US local: +1 864-991-4103 United Kingdom toll-free: +44 808 175 1536 United Kingdom local: +44 1400 220156 Conference ID: 886570
For additional dial-in options, please use this link.
ABOUT GDEV GDEV is a hub of gaming studios, focused on development and growth of its franchise portfolio across various genres and platforms. With a diverse range of subsidiaries including Nexters and Cubic Games among others, GDEV strives to create games that will inspire and engage millions of players for years to come. Its franchises, such as Hero Wars, Island Hoppers, Pixel Gun 3D and others have accumulated hundreds of millions of installs worldwide. For more information, please visit gdev.inc
CONTACTS: Investor Relations Roman Safiyulin | Chief Corporate Development Officer [email protected]
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS Certain statements in this press release may constitute “forward-looking statements” for purposes of the federal securities laws. Such statements are based on current expectations that are subject to risks and uncertainties. In addition, any statements that refer to projections, forecasts or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking statements.
The forward-looking statements contained in this press release are based on the Company’s current expectations and beliefs concerning future developments and their potential effects on the Company. There can be no assurance that future developments affecting the Company will be those that the Company has anticipated. Forward-looking statements involve a number of risks, uncertainties (some of which are beyond the Company’s control) or other assumptions. You should carefully consider the risks and uncertainties described in the “Risk Factors” section of the Company’s 2023 Annual Report on Form 20-F, filed by the Company on April 29, 2024, and other documents filed by the Company from time to time with the Securities and Exchange Commission. Should one or more of these risks or uncertainties materialize, or should any of the Company’s assumptions prove incorrect, actual results may vary in material respects from those projected in these forward-looking statements. Forward-looking statements speak only as of the date they are made. Readers are cautioned not to put undue reliance on forward-looking statements, and the Company undertakes no obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws.
Oral presentation highlighted results from confirmatory Phase 3 RESILIENT study of TNX-102 SL (sublingual cyclobenzaprine HCl) treatment demonstrating statistically significant improvement in primary endpoint of fibromyalgia nociplastic pain and in all six key secondary endpoints, including sleep quality
NDA submission on track for second half 2024; Fast Track designation granted by FDA; FDA decision expected 2025
TNX-102 SL is a potential non-opiod analgesic targeting non-restorative sleep in fibromyalgia: Post hoc analyses highlight strong correlations between improvements in nociplastic pain and sleep quality
Nociplastic pain originates from altered pain perception in the brain and is the type of pain that manifests in fibromyalgia and other chronic overlapping pain conditions (COPCs)
CHATHAM, N.J., Aug. 28, 2024 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP) (Tonix or the Company), a fully-integrated biopharmaceutical company with marketed products and a pipeline of development candidates, presented data in an oral presentation at the 2024 Military Health System Research Symposium (MHSRS), held August 26-29, 2024, in Kissimmee, Fla. A copy of the Company’s presentation, titled “Assuaging Agony: Novel Pain Therapeutics”, is available under the Scientific Presentations tab of the Tonix website at www.tonixpharma.com.
In the Phase 3 RESILIENT study, TNX-102 SL met the pre-specified primary endpoint of significantly reducing daily pain compared to placebo (p-value=0.00005) in participants with fibromyalgia. TNX-102 SL demonstrated broad syndromal benefits with statistically significant improvement in all six pre-specified key secondary endpoints including those related to improving sleep quality, reducing fatigue, and improving patient global ratings and overall fibromyalgia symptoms and function. A post hoc analysis showed strong correlations between improvements in pain and sleep quality at Week 14, supporting the concept that targeting sleep quality has the potential to achieve syndromal improvement in fibromyalgia. TNX-102 SL was well tolerated with an adverse event profile comparable to prior studies and no new safety signals observed.
“Traditional analgesics like NSAIDs or opioids often prove ineffective, if not deleterious, as strategies for treating fibromyalgia,” said Seth Lederman, M.D., Chief Executive Officer of Tonix Pharmaceuticals. “In contrast, TNX-102 SL provided broad-spectrum symptom relief in the Phase 3 RESILIENT study and was designed as a bedtime treatment to target non-restorative sleep and improve sleep quality. With the statistically significant results of two positive Phase 3 studies of TNX-102 SL in fibromyalgia we believe TNX-102 SL has the potential to be the first new treatment option for fibromyalgia patients in 15 years.”
Dr. Lederman continued, “Fibromyalgia is the prototypic nociplastic syndrome and chronic overlapping pain condition (COPC)3,4,5. Our results in fibromyalgia suggest potential for TNX-102 SL in treating other COPCs like post-concussive syndrome6, in which sleep disturbances correlate with persistence and severity. In addition, we expect to begin enrolling this quarter in a trial of TNX-102 SL for acute stress disorder/acute stress reaction in the immediate aftermath of motor vehicle collision in the U.S. Department of Defense (DoD)-funded Optimizing Acute Stress Reaction Interventions (OASIS) trial conducted by the University of North Carolina under an investigator-initiated investigational new drug (IND) application.
Tonix remains on track to submit a new drug application (NDA) to the FDA in the second half of 2024 for TNX-102 SL for the management of fibromyalgia. A decision on approval is expected in 2025.
1Moldofsky H, et al. Psychosom Med. 1975;37:341-51
2Moldofsky H, Scarisbrick P. Psychosom Med. 1976;38:35-44
3Fitzcharles MA, et al. Lancet. 2021;397:2098-110
4Clauw DJ. Ann Rheum Dis. Published Online First: 2024
5Kaplan CM, et al. Nat Rev Neurol. 2024;20, 347–363
6Kureshi S et al. Healthcare (Basel) 2024 12(3): 289.
About Fibromyalgia
Fibromyalgia is a chronic pain disorder that is understood to result from amplified sensory and pain signaling within the central nervous system. Fibromyalgia afflicts more than 10 million adults in the U.S., the majority of whom are women. Symptoms of fibromyalgia include chronic widespread pain, non-restorative sleep, fatigue, and brain fog (or cognitive dysfunction). Other associated symptoms include mood disturbances, including anxiety and depression, headaches, and abdominal pain or cramps. Individuals suffering from fibromyalgia struggle with their daily activities, have impaired quality of life, and frequently are disabled. Physicians and patients report common dissatisfaction with currently marketed products. According to the recent report from the U.S. National Academies of Sciences, fibromyalgia is a diagnosable condition that may also occur in the context of Long COVID
About TNX-102 SL
TNX-102 SL is a centrally acting, non-opioid, non-addictive, bedtime investigational drug. The tablet is a patented sublingual formulation of cyclobenzaprine hydrochloride developed for the management of fibromyalgia. In December 2023, the company announced highly statistically significant and clinically meaningful topline results in RESILIENT, the second pivotal Phase 3 clinical trial of TNX-102 SL for the management of fibromyalgia. In the study, TNX-102 SL met its pre-specified primary endpoint, significantly reducing daily pain compared to placebo (p=0.00005) in participants with fibromyalgia. Statistically significant and clinically meaningful results were also seen in all six key secondary endpoints related to improving sleep quality, reducing fatigue and improving overall fibromyalgia symptoms and function. RELIEF, the first statistically significant Phase 3 trial of TNX-102 SL in fibromyalgia, was completed in December 2020. It met its pre-specified primary endpoint of daily pain reduction compared to placebo (p=0.010) and showed activity in key secondary endpoints. In both pivotal studies, the most common treatment-emergent adverse event was tongue or mouth numbness at the administration site, which was temporally related to dosing, self-limited, never rated as severe, and rarely led to study discontinuation (one participant in each study). TNX-102 SL was recently granted Fast Track Designation by the FDA for the management of fibromyalgia and remains on track to submit an NDA to FDA in the second half of 2024.
About Nociplastic Pain
Nociplastic pain is the third category of pain distinct from nociceptive pain and neuropathic pain. Nociplastic pain is characterized by pain arising from altered nociception despite no evidence of actual or threatened tissue damage causing activation of peripheral nociceptors or somatosensory system disease or lesion. Its underlying pathophysiology involves altered pain processing by the central nervous system (CNS). Nociplastic syndromes, officially recognized by the International Association for the Study of Pain (IASP) in 2017, also include several other chronic overlapping pain conditions: myalgic encephalomyelitis/chronic fatigue syndrome, irritable bowel syndrome, temporomandibular disorders, forms of chronic back pain and chronic headache. The pathophysiology of nociplastic pain involves central sensitization (CS), where neurons of the CNS become hyperexcitable, amplifying pain signals. CS can be triggered by peripheral pain stimuli, emotional stress, or other factors, leading to persistent pain despite no peripheral nociceptive input.
Tonix Pharmaceuticals Holding Corp.*
Tonix is a fully-integrated biopharmaceutical company focused on developing, licensing and commercializing therapeutics to treat and prevent human disease and alleviate suffering. Tonix recently announced the U.S. DoD, Defense Threat Reduction Agency (DTRA) awarded it a contract for up to $34 million over five years to develop TNX-4200 small molecule broad-spectrum antiviral agents targeting CD45 for the prevention or treatment of infections to improve the medical readiness of military personnel in biological threat environments. Tonix owns and operates a state-of-the art infectious disease research facility in Frederick, MD. The company also owns a Good Manufacutring Practice (GMP)-capable advanced manufacturing facility in Dartmouth, MA, which was purpose-built to manufacture TNX-801, a potential mpox vaccine, and the GMP suites are ready to be reactivated in case of a national emergency. Tonix’s development portfolio is focused on CNS disorders. Tonix’s priority is to submit an NDA to the FDA in the second half of 2024 for TNX-102 SL, a product candidate for which two statistically significant Phase 3 studies have been completed for the management of fibromyalgia. The FDA has granted Fast Track designation to TNX-102 SL for the management of fibromyalgia. TNX-102 SL is also being developed to treat acute stress reaction. Tonix’s CNS portfolio includes TNX-1300 (cocaine esterase), a biologic designed to treat cocaine intoxication that has Breakthrough Therapy designation, which is enrolling in a potential pivotal Phase 2 trial. Tonix’s immunology development portfolio consists of biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases. Tonix also has product candidates in development in the areas of rare disease and infectious disease. Tonix Medicines, our commercial subsidiary, markets Zembrace® SymTouch® (sumatriptan injection) 3 mg and Tosymra® (sumatriptan nasal spray) 10 mg for the treatment of acute migraine with or without aura in adults.
*Tonix’s product development candidates are investigational new drugs or biologics and have not been approved for any indication.
Zembrace SymTouch and Tosymra are registered trademarks of Tonix Medicines. All other marks are property of their respective owners.
This press release and further information about Tonix can be found at www.tonixpharma.com.
Forward Looking Statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; risks related to the failure to successfully market any of our products; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2023, as filed with the Securities and Exchange Commission (the “SEC”) on April 1, 2024, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
MALVERN, Pa., Aug. 28, 2024 (GLOBE NEWSWIRE) — Ocugen, Inc. (“Ocugen” or the “Company”) (NASDAQ: OCGN), a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines, today announced that dosing is complete in the third cohort of its Phase 1/2 GARDian clinical trial for OCU410ST (AAV-hRORA)—a modifier gene therapy candidate being developed for Stargardt disease. Stargardt disease affects approximately 100,000 people in the United States (U.S.) and Europe.
“With all patients dosed in cohort 3 (high dose), Phase 1 of the dose-escalation portion of the trial is complete,” said Dr. Huma Qamar, Chief Medical Officer of Ocugen. “We will continue to advance the trial as efficiently as possible, and work toward fulfilling an unmet medical need for Stargardt patients.”
Three subjects received a single subretinal injection of the highest dose (2.25×1011 vg/mL) being tested. The GARDian clinical trial is being performed at six leading retinal surgery centers across the U.S.
“OCU410ST is a novel modifier gene therapy that has the potential to be a one-time treatment given by subretinal injection in the operating room,” said Charles Wykoff, MD, PhD, Director of Research, Retina Consultants of Texas, lead investigator in the study. “The safety and tolerability profile of OCU410ST remains encouraging as the clinical trial progresses and brings hope to patients with Stargardt disease, who have no FDA-approved treatment options.”
The GARDian clinical trial will assess the safety and efficacy of unilateral subretinal administration of OCU410ST in subjects with Stargardt disease and will be conducted in two phases. Phase 1 is a multicenter, open-label, dose-ranging study consisting of three dose levels [low dose (3.75×1010 vg/mL), medium dose (7.5×1010 vg/mL), and high dose (2.25×1011 vg/mL)].
Ocugen remains committed to advancing treatments for blindness, focusing on innovative gene therapy solutions that aim to provide lasting benefits to patients.
The Company will continue to provide clinical updates on a periodic basis.
About Stargardt Disease Stargardt disease is a genetic eye disorder that causes retinal degeneration and vision loss. Stargardt disease is the most common form of inherited macular degeneration. The progressive vision loss associated with Stargardt disease is caused by the degeneration of photoreceptor cells in the central portion of the retina called the macula.
Decreased central vision due to loss of photoreceptors in the macula is the hallmark of Stargardt disease. Some peripheral vision is usually preserved. Stargardt disease typically develops during childhood or adolescence, but the age of onset and rate of progression can vary. The retinal pigment epithelium (RPE), a layer of cells supporting photoreceptors, is also affected in people with Stargardt disease.
About OCU410ST OCU410ST utilizes an AAV delivery platform for the retinal delivery of the RORA (RAR Related Orphan Receptor A) gene. It represents Ocugen’s modifier gene therapy approach, which is based on Nuclear Hormone Receptor (NHR) RORA that regulates pathway links to Stargardt disease such as lipofuscin formation, oxidative stress, compliment formation, inflammation, and cell survival networks.
About Ocugen, Inc. Ocugen, Inc. is a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines that improve health and offer hope for patients across the globe. We are making an impact on patients’ lives through courageous innovation—forging new scientific paths that harness our unique intellectual and human capital. Our breakthrough modifier gene therapy platform has the potential to treat multiple retinal diseases with a single product, and we are advancing research in infectious diseases to support public health and orthopedic diseases to address unmet medical needs. Discover more at www.ocugen.com and follow us on X and LinkedIn.
Forward-Looking Statements This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995, including, but not limited to, statements regarding qualitative assessments of available data, potential benefits, expectations for ongoing clinical trials, anticipated regulatory filings and anticipated development timelines, which are subject to risks and uncertainties. We may, in some cases, use terms such as “predicts,” “believes,” “potential,” “proposed,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Such statements are subject to numerous important factors, risks, and uncertainties that may cause actual events or results to differ materially from our current expectations, including, but not limited to, the risks that preliminary, interim and top-line clinical trial results may not be indicative of, and may differ from, final clinical data; that unfavorable new clinical trial data may emerge in ongoing clinical trials or through further analyses of existing clinical trial data; that earlier non-clinical and clinical data and testing of may not be predictive of the results or success of later clinical trials; and that that clinical trial data are subject to differing interpretations and assessments, including by regulatory authorities. These and other risks and uncertainties are more fully described in our periodic filings with the Securities and Exchange Commission (SEC), including the risk factors described in the section entitled “Risk Factors” in the quarterly and annual reports that we file with the SEC. Any forward-looking statements that we make in this press release speak only as of the date of this press release. Except as required by law, we assume no obligation to update forward-looking statements contained in this press release whether as a result of new information, future events, or otherwise, after the date of this press release.
RICHMOND, Va.–(BUSINESS WIRE)– Bowlero Corp. (NYSE: BOWL) (“Bowlero” or the “Company”), one of the world’s premier operators of location-based entertainment, will report financial results for the fourth quarter and full year fiscal 2024 on Thursday, September 5, 2024, after the U.S. stock market closes. Management will discuss the results via webcast at 4:30 PM ET on the same day.
The live webcast, replay, and results presentation will be available in the Events & Presentations section of the Bowlero Investor Relations website at https://ir.bowlerocorp.com/ .
About Bowlero Corp.
Bowlero Corporation is one of the world’s premier operators of location-based entertainment. With approximately 350 locations across North America, the Company serves more than 40 million guest visits annually through a family of brands that include Lucky Strike, Bowlero and AMF. In 2019, Bowlero acquired the Professional Bowlers Association, the major league of bowling and a growing media property that boasts millions of fans around the globe. For more information on Bowlero, please visit BowleroCorp.com.
MALVERN, Pa., Aug. 26, 2024 (GLOBE NEWSWIRE) — Ocugen, Inc. (“Ocugen” or the “Company”) (NASDAQ: OCGN), a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines, today announced that Health Canada provided a “No Objection Letter” to initiate the OCU400 Phase 3 liMeliGhT (pronounced “limelight”) clinical trial in Canada. OCU400 is a modifier gene therapy product candidate being developed for retinitis pigmentosa (RP).
“Expanding the clinical trial to Canada is significant as it will provide an opportunity to reach a broader patient population encompassing many gene mutations associated with RP,” said Dr. Shankar Musunuri, Chairman, CEO, and Co-Founder of Ocugen. “The Health Canada trial will run in parallel with the U.S. FDA trial, expediting the ability to potentially provide a gene-agnostic treatment option to approximately 110,000 patients in the United States (U.S.) and Canada.”
Currently there are approximately 10,000 patients in Canada with RP and 1.6 million patients globally. The Phase 3 study in Canada will enroll up to 50 subjects across a maximum of 5 sites for the liMeliGhT clinical trial.
Over 200 mutations in more than 100 genes have been linked to RP. The Phase 3 study, spanning one year, will enroll 150 participants divided into two study arms: 75 participants with RHO gene mutations and 75 participants who are gene agnostic. In each arm, participants will be randomized in a 2:1 ratio to receive either treatment (2.5 x 1010 vg/eye of OCU400) or remain in an untreated control group, respectively. The liMeliGhT study is recruiting patients aged eight and older, covering the full spectrum from early to late stages of RP progression.
An enhanced sensitive and specific measurement of functional vision test—Luminance Dependent Navigation Assessment (LDNA)—is the primary endpoint for the study. Specifically, the primary endpoint is a measurement of the change in functional vision from baseline to week 52 as measured by the ability of a study participant to navigate through a maze (the LDNA). Those who demonstrate an improved ability to navigate the maze in dimmer light (i.e., by ≥2 Lux levels) compared to baseline will be classified as “responders” to the therapy. The liMeliGhT study will focus on the proportion of responders in both the treated and untreated eyes.
“Establishing clinical sites in Canada may expedite recruitment and open doors for broader commercialization with the U.S. and Europe,” said Dr. Huma Qamar, Chief Medical Officer at Ocugen. “With only one currently approved treatment targeting a single mutation associated with RP, there remains a significant unmet medical need, and patients worldwide are eager for new therapeutic options. It is highly rewarding to extend our efforts into a new region and offer hope to Canadian patients with RP.”
Ocugen previously announced that OCU400 has received orphan drug and RMAT designations from the FDA. OCU400 remains on track for the 2026 BLA and MAA approval targets.
About OCU400
OCU400 is the Company’s modifier gene therapy product based on a nuclear hormone receptor gene called NR2E3. This gene regulates diverse physiological functions within the retina, such as photoreceptor development and maintenance, metabolism, phototransduction, inflammation, and cell survival. Retinal cells in RP patients have a dysfunctional gene network, and OCU400 resets this network to reestablish a healthy cellular homeostasis—which has the potential to improve vision in patients with RP.
About RP
RP is a group of rare genetic disorders that cause a breakdown in the cells of the retina, leading to vision loss and blindness. RP is associated with mutations in more than 100 genes.
There are no approved treatment options that slow or stop the progression of multiple forms of RP. Proposed treatments for RP include gene replacement therapy, retinal implant devices, retinal transplantation, stem cells, vitamin therapy, and other pharmacological treatments. Current gene replacement therapies are promising but are limited to treating just a single mutation. In addition, while gene therapies may provide a new functional gene, they do not necessarily eliminate the underlying genetic defect, which may still cause stress and toxic effects leading to retinal degeneration. Therefore, the development of gene-specific replacement therapy will not address all forms of RP, especially when multiple and unknown genes are involved. Thus, novel therapeutic approaches targeting the broader RP disease in a gene-agnostic manner offer greater hope for patients.
About Ocugen, Inc.
Ocugen, Inc. is a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies, biologics, and vaccines that improve health and offer hope for patients across the globe. We are making an impact on patients’ lives through courageous innovation—forging new scientific paths that harness our unique intellectual and human capital. Our breakthrough modifier gene therapy platform has the potential to treat multiple retinal diseases with a single product, and we are advancing research in infectious diseases to support public health and orthopedic diseases to address unmet medical needs. Discover more at www.ocugen.com and follow us on X and LinkedIn.
Forward-Looking Statements
This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995, including, but not limited to, statements regarding qualitative assessments of available data, potential benefits, expectations for ongoing clinical trials, anticipated regulatory filings and anticipated development timelines, which are subject to risks and uncertainties. We may, in some cases, use terms such as “predicts,” “believes,” “potential,” “proposed,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Such statements are subject to numerous important factors, risks, and uncertainties that may cause actual events or results to differ materially from our current expectations, including, but not limited to, the risks that preliminary, interim and top-line clinical trial results may not be indicative of, and may differ from, final clinical data; that unfavorable new clinical trial data may emerge in ongoing clinical trials or through further analyses of existing clinical trial data; that earlier non-clinical and clinical data and testing of may not be predictive of the results or success of later clinical trials; and that that clinical trial data are subject to differing interpretations and assessments, including by regulatory authorities. These and other risks and uncertainties are more fully described in our periodic filings with the Securities and Exchange Commission (SEC), including the risk factors described in the section entitled “Risk Factors” in the quarterly and annual reports that we file with the SEC. Any forward-looking statements that we make in this press release speak only as of the date of this press release. Except as required by law, we assume no obligation to update forward-looking statements contained in this press release whether as a result of new information, future events, or otherwise, after the date of this press release.