Baudax Bio is a pharmaceutical company focused on innovative products for acute care settings. ANJESO is the first and only 24-hour, intravenous (IV) COX-2 preferential non-steroidal anti-inflammatory (NSAID) for the management of moderate to severe pain. In addition to ANJESO, Baudax Bio has a pipeline of other innovative pharmaceutical assets including two novel neuromuscular blocking agents (NMBs) and a proprietary chemical reversal agent specific to these NMBs. For more information, please visit www.baudaxbio.com.
Gregory Aurand, Senior Research Analyst, Healthcare Services & Medical Devices, Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Baudax reported 3Q 2022 results. 3Q revenues reported of $0.238 million were well below our expectation of $0.965 million. While vials sold increased year-over-year approximately 16%, much of the 3Q volume was discounted into 340B hospitals causing revenues to decline 15% from the prior year.
Feeling the pain. In the Covid environment that doesn’t seem to go away, hospitals face financial pressures as they struggle to curtail costs. For a single product company like Baudax Bio, the hospital issues have inflicted continued pain that is difficult to overcome. Hospitals look for less expensive alternatives in pain management. Recognizing that Covid-related issues facing hospitals now could continue into and through 2023, the Company eliminated the remaining commercial team in September 2022.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Revenue Increased 36% to $43.2 Million Compared to $31.8 Million in Q3 2021 Nine Month Revenue Increased 46% to $119.2 Million Compared to $81.9 Million
Adjusted EBITDA of $15.9 Million, 36.7% of Revenue Nine Month Adjusted EBITDA of $38.7 Million, 32.5% of Revenue
DENVER, Nov. 9, 2022 /CNW/ – Medicine Man Technologies Inc. operating as Schwazze, (OTCQX: SHWZ) (NEO: SHWZ) (“Schwazze” or the “Company”), today announced financial results for the third quarter ended September 30, 2022 (“Q3 2022”).
Q3 2022 Financial Summary:
Revenues of $43.2 million increased 36% compared to $31.8 million in quarter ended September 30, 2021 (“Q3 2021”)
Retail sales were $39.8 million up 92% to $20.7 million when compared to Q3 2021
Gross Margin of $26.0 million, 60.1% of revenue, compared to $15.1 million and 47.3% of revenue in Q3 2021
Net Income was $1.8 million compared to a Net Income of $1.0 million for the same period last year
Adjusted EBITDA of $15.9 million was 36.7% of revenue, compared to $8.8 million for the same period last year
Colorado two year stacked IDs for Q3 2022 compared to Q3 2022 and Q3 2020 for same store sales(1) were (9.7%) and one year IDs(1) were (10.6%) comparing Q3 2022 to Q3 2021
Average basket size (1) for Q3 2022 was $60.96 up slightly by 0.1% compared to Q3 2021
Recorded customer visits (1) for Q3 2022 totaled 452,220 down 10.7%, compared to Q3 2021
New Mexico two year stacked IDs for Q3 2022 compared to Q3 2021 and Q3 2020 for same store sales(1) were 52.9% and one year IDs(1) were 48.4% comparing Q3 2022 to Q3 2021
Average basket size (1) for Q3 2022 was $52.67 down 12.2% compared to Q3 2021
Recorded customer visits (1) for Q3 2022 totaled 231,137 up 69.0%, compared to Q3 2021
Corporate Update: Since December 2021, Schwazze has closed acquisitions adding 15 cannabis dispensaries, 10 in New Mexico and five in Colorado as well as four cultivation facilities in New Mexico and one in Colorado and one manufacturing asset in New Mexico. This year Schwazze has opened two new dispensaries in New Mexico. This brings our total dispensary count to 35 between Colorado and New Mexico.
Justin Dye, Chairman and CEO of Schwazze stated, “I am proud of the entire Schwazze team, and I would like to thank them for their hard work this past quarter and year. Despite a challenging economic backdrop, we outperformed our markets in Colorado by 12%. We’ve worked hard to continue to grow our market share, increase our profitability rate and generate free cash flow from operations, after paying taxes and CAPEX, placing us in an exclusive club within the cannabis sector. This is a proof point that we are well on our way to building Schwazze into a unique regional powerhouse. I believe our distinctive operating capabilities, applied to attractive growth opportunities within our sector, will reward our shareholders with attractive risk adjusted returns. The potential of favorable regulatory reform in the near-term would obviously accelerate and amplify those returns.”
Q3 2022 Revenue Revenues for the three months ended September 30, 2022 totaled $43,190,986, including (i) retail sales of $39,759,734 (ii) wholesale sales of $3,335,252 and (iii) other operating revenues of $96,000, compared to revenues of $31,835,305, including (i) retail sales of $20,741,864, (ii) wholesale sales of $11,022,519, and (iii) other operating revenues of $70,922 during the three months ended September 30, 2021, representing an increase of $11,355,681 or 36%. The most influential factor driving revenue increases in the third quarter of 2022 as compared to the same period in 2021 is acquisition activity. Revenue for the quarter ended September 30, 2022 included revenue from four consummated acquisitions in Colorado and revenue from the Company’s initial entrance into the New Mexico market with the acquisition of R. Greenleaf, which were not in revenue for the same period in 2021. Revenue from wholesale sales decreased, due in large part to continued pricing pressure in the Colorado wholesale market as a result of supply saturation in flower and bulk distillate products.
Cost of goods and services for the three months ended September 30, 2022, totaled $17,226,451 compared to cost of goods and services of $16,779,313 during the three months ended September 30, 2021, representing an increase of $447,138 or 3%. Overall cost of goods and services increased due to the same acquisition activities that generated substantial increases in revenue, but the rate at which cost of goods and services increases from acquisition activity occurs at a lower rate than increases in revenue from acquisition activity due to lower wholesale flower pricing in Colorado and substantial vertical integration in New Mexico and increased retail revenue, which has better gross margin, as a percentage of the total revenue.
Gross profit was $25,964,535 million dollars for the quarter compared to $15,055,992 during the same period in 2021. Gross profit margin increased as a percentage of revenue from 47.3% to 60.1%. This positive result reflects a higher percentage of retail sales, our consolidated purchasing approach, the implementation of our retail playbook, and vertical product sales in New Mexico.
Operating expenses for the quarter, totaled $14,849,677, compared to operating expenses of $11,218,992 during the same quarter 2021, representing an increase of $3,630,685 or 32%. This increase is due to increased selling, general and administrative expenses, professional service fees, salaries, benefits and related employment costs driven by growth from acquisitions offset by stock-based compensation.
Other expense, net for the three months ended September 30, 2022 totaled $3,712,108 compared to $1,555,427 during the three months ended September 30, 2021, representing an increase in other expense of $2,156,681 or 139%. The increase in other expenses is due to higher interest payments due on the Company’s debt obligations as a result of compounding interest with the passage of time and higher debt balances, which was partially offset this quarter by the revaluation of the derivative liability related to the Investor Notes issued in December 2021 that was recognized as income in the three months ended September 30, 2022.
Adjusted EBITDA for Q3 2022 was $15,860,466 representing 36.7% of revenue, compared to $8,797,641 and 27.6% of revenue for the same period last year. This is derived from Operating Income and adjusting one-time expenses, merger and acquisition and capital raising costs, non-cash related compensation costs, and depreciation and amortization. See the financial table for Adjusted EBITDA below adjustment for details.
For nine months ending September 30, 2022, the Company used cash for operations of $3,957,263 compared to generating cash of $4,814,104 for the same period in 2021. The Company has cash and cash equivalents of $38.7 million at the end of Q3 2022.
Nancy Huber, CFO for Schwazze commented, “During the third quarter we continued our focus on reducing operating and SG&A expenses. Our third quarter gross margin and operating expenses improved over the second quarter in both dollars and percent of revenue. Our balance sheet remains strong, with ample liquidity. We continue to be committed to delivering positive cash flow before acquisition costs for the year while driving organic growth with the opening of two stores in New Mexico in the third quarter.”
2022 Guidance The Company is providing guidance for the fiscal year. FY 2022 revenue is projected to be $155 million to $165 million, and the FY 2022 adjusted EBITDA is projected to be from $51 million to $56 million. We are on target to deliver the lower end of the range for adjusted EBITDA which was a fourth quarter annualized run-rate of $60-72 million dollars. We expect to be slightly below the projected revenues which was a fourth quarter annualized run-rate of $175 million to $200 million. This lower-than-expected revenue in Q4 is due to lower than expected wholesale sales, and construction delays in new store openings in New Mexico.
The company generated $4 million in cash from operations in the third quarter and expects to generate positive cash flow before acquisitions for the year.
NOTES:
(1)
Schwazze did not own all the assets and entities in part of 2021, 2020 and 2019 and is using unaudited numbers for this comparison.
Adjusted EBITDA represents income (loss) from operations, as reported, before tax, adjusted to exclude non-recurring items, other non-cash items, including stock-based compensation expense, depreciation, and amortization, and further adjusted to remove acquisition and capital raise related costs, and other one-time expenses, such as severance, retention, and employee relocation. The Company uses adjusted EBITDA as it believes it better explains the results of its core business. The Company has not reconciled guidance for adjusted EBITDA to the corresponding GAAP financial measure because it cannot provide guidance for the various reconciling items. The Company is unable to provide guidance for these reconciling items because it cannot determine their probable significance, as certain items are outside of its control and cannot be reasonably predicted. Accordingly, a reconciliation to the corresponding GAAP financial measure is not available without unreasonable effort.
Webcast – November 9, 2022 – 5:00 PM EDT Investors and stakeholders may participate in the conference call by dialing 416-764-8650 or by dialing North American toll free 1-888-664-6383 or listen to the webcast from the Company’s website at https://ir.schwazze.com The webcast will be available on the Company’s website and on replay until November 16, 2022, and may be accessed by dialing 1-888-390-0541 / 997573 #.
Following their prepared remarks, Chief Executive Officer, Justin Dye; President, Nirup Krishnamurthy; and Chief Financial Officer, Nancy Huber will answer investor questions. Investors may submit questions in advance or during the conference call itself through the weblink: https://app.webinar.net/x0q6rpnP84n. This weblink has been posted to the Company’s website and will be archived on the website. All Company SEC filings can also be accessed on the Company website at https://ir.schwazze.com/sec-filings
About Schwazze Schwazze (OTCQX: SHWZ, NEO: SHWZ) is building a premier vertically integrated regional cannabis company with assets in Colorado and New Mexico and will continue to take its operating system to other states where it can develop a differentiated regional leadership position. Schwazze is the parent company of a portfolio of leading cannabis businesses and brands spanning seed to sale. The Company is committed to unlocking the full potential of the cannabis plant to improve the human condition. Schwazze is anchored by a high-performance culture that combines customer-centric thinking and data science to test, measure, and drive decisions and outcomes. The Company’s leadership team has deep expertise in retailing, wholesaling, and building consumer brands at Fortune 500 companies as well as in the cannabis sector. Schwazze is passionate about making a difference in our communities, promoting diversity and inclusion, and doing our part to incorporate climate-conscious practices. Medicine Man Technologies, Inc. was Schwazze’s former operating trade name. The corporate entity continues to be named Medicine Man Technologies, Inc. Schwazze derives its name from the pruning technique of a cannabis plant to enhance plant structure and promote healthy growth.
Forward-Looking Statements Such forward-looking statements may be preceded by the words “plan,” “will,” “may,” “continue,” “anticipate,” “become,” “build,” “develop,” “expect,” “believe,” “poised,” “project,” “approximate,” “could,” “potential,” or similar expressions as they relate to Schwazze. Forward-looking statements include the guidance provided regarding the Company’s Q4 2022 performance and annual capital spending. Forward-looking statements are not guarantees of future events or performance, are based on certain assumptions, and are subject to various known and unknown risks and uncertainties, many of which are beyond the Company’s control and cannot be predicted or quantified. Consequently, actual events and results may differ materially from those expressed or implied by such forward-looking statements. Such risks and uncertainties include, without limitation, risks and uncertainties associated with (i) our inability to manufacture our products and product candidates on a commercial scale on our own or in collaboration with third parties; (ii) difficulties in obtaining financing on commercially reasonable terms; (iii) changes in the size and nature of our competition; (iv) loss of one or more key executives or scientists; (v) difficulties in securing regulatory approval to market our products and product candidates; (vi) our ability to successfully execute our growth strategy in Colorado and New Mexico and outside the states, (vii) our ability to identify and consummate future acquisitions that meet our criteria, (viii) our ability to successfully integrate acquired businesses and realize synergies therefrom, (ix) the ongoing COVID-19 pandemic, * the timing and extent of governmental stimulus programs, (xi) the uncertainty in the application of federal, state and local laws to our business, and any changes in such laws, and (xii) our ability to achieve the target metrics, including our annualized revenue and EBIDTA run rates set out in our Q4 2022 guidance. More detailed information about the Company and the risk factors that may affect the realization of forward-looking statements is set forth in the Company’s filings with the Securities and Exchange Commission (SEC), including the Company’s Annual Report on Form 10-K and its Quarterly Reports on Form 10-Q. Investors and security holders are urged to read these documents free of charge on the SEC’s website at http://www.sec.gov. The Company assumes no obligation to publicly update or revise its forward-looking statements as a result of new information, future events or otherwise except as required by law.
SASKATOON, Saskatchewan, Canada, November 9, 2022 – MustGrow Biologics Corp. (CSE: MGRO; OTCQB: MGROF; FRA: 0C0) (the “Company” or “MustGrow”), is pleased to announce that the TSX Venture Exchange (“TSXV“) has approved the Company’s application to list its common shares (the “Shares“) on the TSXV. The Shares will commence trading on the TSXV under the ticker symbol “MGRO” at the opening of the Market on November 11, 2022. Shareholders will not be required to take any action in connection with MustGrow’s listing on the TSXV. The Shares will also continue to be listed on the OTCQB Marketplace in the United States under the symbol “MGROF” and on the Frankfurt Stock Exchange under the symbol “0C0”.
In connection with the listing of the Shares on the TSXV, the Company has submitted a request to voluntarily delist the Shares from the Canadian Securities Exchange (“CSE“). The last day of trading of the Company’s shares on the CSE will be November 10, 2022.
For further details, please refer to the Listing Application on the Company’s SEDAR profile available at www.sedar.com.
———
About MustGrow
MustGrow is an agriculture biotech company developing organic biopesticides and bioherbicides by harnessing the natural defense mechanism of the mustard plant to protect the global food supply from diseases, insect pests, and weeds. MustGrow and its leading global partners – Janssen PMP (pharmaceutical division of Johnson & Johnson), Bayer, Sumitomo Corporation, and Univar Solutions’ NexusBioAg – are developing mustard-based organic solutions to potentially replace harmful synthetic chemicals. Over 150 independent tests have been completed, validating MustGrow’s safe and effective approach to crop and food protection. Pending regulatory approval, MustGrow’s patented liquid products could be applied through injection, standard drip, or spray equipment, improving functionality and performance features. Now a platform technology, MustGrow and its global partners are pursuing applications in several different industries from preplant soil treatment and weed control, to postharvest disease control and food preservation. MustGrow has approximately 49.7 million basic common shares issued and outstanding and 55.6 million shares fully diluted. For further details, please visit www.mustgrow.ca.
Certain statements included in this news release constitute “forward-looking statements” which involve known and unknown risks, uncertainties and other factors that may affect the results, performance or achievements of MustGrow.
Generally, forward-looking information can be identified by the use of forward-looking terminology such as “plans”, “expects”, “is expected”, “budget”, “estimates”, “intends”, “anticipates” or “does not anticipate”, or “believes”, or variations of such words and phrases or statements that certain actions, events or results “may”, “could”, “would”, “might”, “occur” or “be achieved”. Examples of forward-looking statements in this news release include statements MustGrow makes regarding the anticipated date of the commencement of trading of the Company’s Shares on the TSXV.
Forward-looking statements are subject to a number of risks and uncertainties that may cause the actual results of MustGrow to differ materially from those discussed in such forward-looking statements, and even if such actual results are realized or substantially realized, there can be no assurance that they will have the expected consequences to, or effects on, MustGrow. Important factors that could cause MustGrow’s actual results and financial condition to differ materially from those indicated in the forward-looking statements include, among others, the following: (i) the preferences and choices of agricultural regulators with respect to product approval timelines; (ii) the ability of MustGrow’s partners to meet obligations under their respective agreements; and (iii) other risks described in more detail in MustGrow’s Annual Information Form for the year ended December 31, 2021 and other continuous disclosure documents filed by MustGrow with the applicable securities regulatory authorities which are available at www.sedar.com. Readers are referred to such documents for more detailed information about MustGrow, which is subject to the qualifications, assumptions and notes set forth therein.
This release does not constitute an offer for sale of, nor a solicitation for offers to buy, any securities in the United States.
Neither the CSE, the TSXV, nor their Regulation Services Provider (as that term is defined in the policies of the CSE and TSXV), nor the OTC Markets has approved the contents of this release or accepts responsibility for the adequacy or accuracy of this release.
CHATHAM, N.J., Nov. 09, 2022 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP), a clinical-stage biopharmaceutical company, today announced a poster presentation by an academic collaborator at Neuroscience 2022 by the Society for Neuroscience being held November 12-16, 2022, at the San Diego Convention Center in San Diego, Calif.
Poster Presentation Details:
Title:
In Vitro Impact of Oxytocin on Human Sensory Neurons
Session:
Poster Session I
Date:
November 14, 2022
Time:
1:00 p.m. – 2:00 p.m. PT
Authors:
David C. Yeomans, Ph.D., Associate Professor, Anesthesiology, Perioperative and Pain Medicine, Stanford University and David Hsu, Ph.D., Senior Scientist, Tonix Pharmaceuticals
Presenter:
David C. Yeomans, Ph.D., Associate Professor, Anesthesiology, Perioperative and Pain Medicine, Stanford University
Tonix is a clinical-stage biopharmaceutical company focused on discovering, licensing, acquiring and developing therapeutics to treat and prevent human disease and alleviate suffering. Tonix’s portfolio is composed of central nervous system (CNS), rare disease, immunology and infectious disease product candidates. Tonix’s CNS portfolio includes both small molecules and biologics to treat pain, neurologic, psychiatric and addiction conditions. Tonix’s lead CNS candidate, TNX-102 SL (cyclobenzaprine HCl sublingual tablet), is in mid-Phase 3 development for the management of fibromyalgia with a new Phase 3 study launched in the second quarter of 2022 and interim data expected in the second quarter of 2023. TNX-102 SL is also being developed to treat Long COVID, a chronic post-acute COVID-19 condition. Tonix initiated a Phase 2 study in Long COVID in the third quarter of 2022 and expects interim data in the second quarter of 2023. TNX-1300 (cocaine esterase) is a biologic designed to treat cocaine intoxication and has been granted Breakthrough Therapy designation by the FDA. A Phase 2 study of TNX-1300 is expected to be initiated in the first quarter of 2023. TNX-1900 (intranasal potentiated oxytocin), a small molecule in development for chronic migraine, is expected to enter the clinic with a Phase 2 study in the fourth quarter of 2022. TNX-601 ER (tianeptine hemioxalate extended-release tablets) is a once-daily formulation of tianeptine being developed as a potential treatment for major depressive disorder (MDD) with a Phase 2 study expected to be initiated in the first quarter of 2023. Tonix’s rare disease portfolio includes TNX-2900 (intranasal potentiated oxytocin) for the treatment of Prader-Willi syndrome. TNX-2900 has been granted Orphan Drug designation by the FDA. Tonix’s immunology portfolio includes biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft and xenograft rejection and for the treatment of autoimmune diseases. A Phase 1 study of TNX-1500 is expected to be initiated in the first half of 2023. Tonix’s infectious disease pipeline consists of a vaccine in development to prevent smallpox and monkeypox, next-generation vaccines to prevent COVID-19, and a platform to make fully human monoclonal antibodies to treat COVID-19. TNX-801, Tonix’s vaccine in development to prevent smallpox and monkeypox, also serves as the live virus vaccine platform or recombinant pox vaccine (RPV) platform for other infectious diseases. A Phase 1 study of TNX-801 is expected to be initiated in Kenya in the first half of 2023. Tonix’s lead vaccine candidate for COVID-19 is TNX-1850, a live virus vaccines based on Tonix’s recombinant pox live virus vector vaccine platform.
*All of Tonix’s product candidates are investigational new drugs or biologics and have not been approved for any indication.
This press release and further information about Tonix can be found at www.tonixpharma.com.
Forward Looking Statements Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; delays and uncertainties caused by the global COVID-19 pandemic; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2021, as filed with the Securities and Exchange Commission (the “SEC”) on March 14, 2022, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
Ocugen, Inc. is a biotechnology company focused on developing and commercializing novel gene therapies, biologicals, and vaccines. The lead product, Covaxin, is a killed-virus vaccine for COVID-19 in-licensed from Bharat Biotech (India). The lead product in its gene therapy program, OCU400, is in Phase 1/2 clinical trials for retinitis pigmentosa.
Robert LeBoyer, Vice President, Research Analyst, Life Sciences , Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
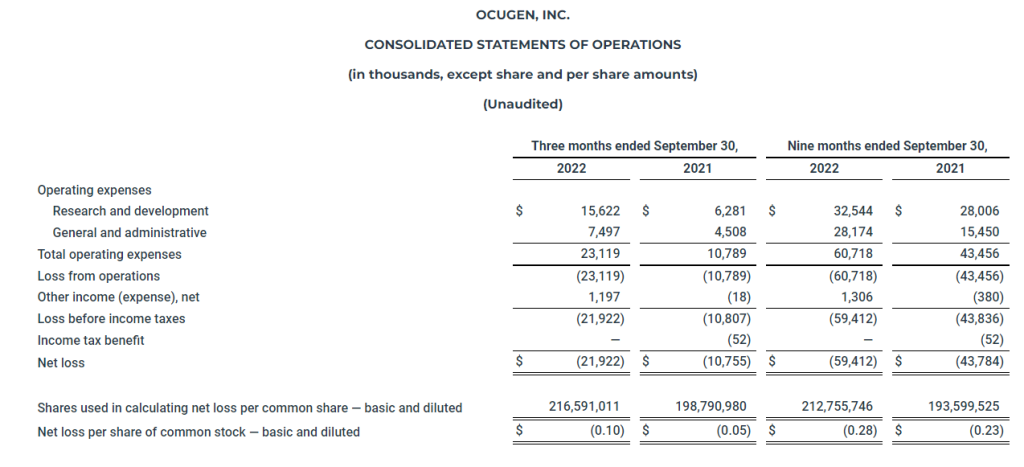
Financial Results Met Expectations. Ocugen reported a 3Q loss of $21.9 million or $(0.10) per share, close to our estimates of $21.2 million and $(0.10) for the quarter. Cash on hand at September 30, 2022 was $101.6 million, which the company expects to last through 2023.
COVID-19 Programs Are Moving Forward. The Phase 2/3 immuno-bridging study for COVAXIN has completed enrollment with data release expected in early 2023. This study is testing COVAXIN to determine the immune response in the current US population, providing a “bridge” for comparability with the Phase 3 trial conducted in India. The study enrolled patients who were unvaccinated and patients that received mRNA vaccines to determine both initial and booster efficacy. Once completed, Ocugen plans to work with government agencies for support and funding for regulatory approval of its COVID-19 products.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Initiated dosing in the third and final cohort of U.S. Phase 1/2 OCU400 gene therapy clinical trial
Expanded product pipeline with OCU500—Ocugen’s mucosal COVID-19 vaccine and OCU410ST for Stargardt disease
Completed enrollment of U.S. Phase 2/3 COVAXIN™ (BBV152) clinical trial
MALVERN, Pa., Nov. 08, 2022 (GLOBE NEWSWIRE) — Ocugen, Inc. (Ocugen or the Company) (NASDAQ: OCGN), a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines, today reported financial results for the quarter ended September 30, 2022, and provided a general business update.
“We achieved several important milestones in the third quarter of 2022,” said Dr. Shankar Musunuri, Chairman, Chief Executive Officer, and Co-Founder of Ocugen. “We expanded our vaccines pipeline by adding a second asset to combat COVID-19, OCU500, through our exclusive license agreement with Washington University to develop, manufacture, and commercialize a mucosal vaccine in the United States, Europe, and Japan.”
“Our modifier gene therapy platform has significant potential to address multiple blindness diseases,” said Dr. Musunuri. “The OCU400 clinical trial is on track, and we are also pleased to announce the addition of OCU410ST to our pipeline as a potential therapy for Stargardt disease—an orphan disease.”
“We continue to deliver on our near-term commitments as we advance our longer-term strategy and goal of bringing solutions to patients with debilitating diseases for whom no appropriate treatment options exist. We are passionate about this goal and anticipate achieving multiple milestones across our programs next year,” Dr. Musunuri concluded.
Business Updates
Vaccines
COVAXIN™ – enrollment was completed, and dosing continues, in the Phase 2/3 immuno-bridging and broadening clinical trial. No safety concerns have been identified to date and efficacy is being continuously monitored. Top line data is expected in early 2023.
OCU500 – a novel adenovirus-vectored mucosal vaccine, specifically designed to block COVID-19 infection at the portal of virus entry and that could prevent transmission as well as provide protection against new variants. This approach represents a potential universal booster, regardless of previous COVID-19 vaccination. Obtaining mucosal immunity has been published as a potential way to prevent infection and transmission, thus limiting the origin of new variants. Mucosal vaccines similar to the Company’s approach are already authorized in China and India. Ocugen intends to work closely with government agencies tasked with pandemic preparedness and response to initiate clinical trials.
Gene Therapies
OCU400
Dosing of subjects with NR2E3 and RHO-related retinitis pigmentosa in Cohort 2 was completed. Based on a review of safety data by the independent Data and Safety Monitoring Board for the clinical trial, dosing has begun in Cohort 3, and enrollment is expected to be completed by the end of the year.
The current clinical trial will also start enrolling patients with Leber congenital amaurosis associated with CEP290 mutations.
OCU410 and OCU410ST – Filings of Investigational New Drug (IND) applications for both dry age-related macular degeneration and Stargardt disease are planned for Q2 2023.
Biologicals
OCU200 – Ocugen is currently executing IND-enabling studies. The filing of an IND application targeting DME is planned for Q1 2023.
Cell Therapies
NeoCart® – Ocugen continues to work with the U.S. Food and Drug Administration to finalize the Phase 3 protocol necessary for the clinical development program of NeoCart®. Ocugen is building its own manufacturing suites to prepare for a NeoCart® clinical trial and as part of an overall research and development expansion.
Third Quarter 2022 Financial Results
The Company’s cash, cash equivalents, and restricted cash totaled $101.6 million as of September 30, 2022, compared to $95.1 million as of December 31, 2021. The Company believes that its current cash and cash equivalents balance will enable it to fund its operations into Q4 2023. The Company had 216.7 million shares of common stock outstanding as of September 30, 2022.
Research and development expenses for the three months ended September 30, 2022, were $15.6 million compared to $6.3 million for the three months ended September 30, 2021. General and administrative expenses for the three months ended September 30, 2022, were $7.5 million compared to $4.5 million for the three months ended September 30, 2021.
Ocugen reported a $0.10 net loss per share for the three months ended September 30, 2022, compared to a $0.05 net loss per share for the three months ended September 30, 2021.
Conference Call and Webcast Details Ocugen has scheduled a conference call and webcast for 8:30 a.m. ET today to discuss the financial results and recent business highlights. Ocugen’s senior management team will host the call, which will be open to all listeners. There will also be a question-and-answer session following the prepared remarks.
Attendees are invited to participate on the call or webcast using the following details:
Dial-in Numbers: (800) 715-9871 for U.S. callers and (646) 307-1963 for international callers
A replay of the call and archived webcast will be available for approximately 45 days following the event on the Ocugen investor site
About Ocugen, Inc. Ocugen, Inc. is a biotechnology company focused on discovering, developing, and commercializing novel gene and cell therapies and vaccines that improve health and offer hope for patients across the globe. We are making an impact on patient’s lives through courageous innovation—forging new scientific paths that harness our unique intellectual and human capital. Our breakthrough modifier gene therapy platform has the potential to treat multiple retinal diseases with a single product, and we are advancing research in infectious diseases to support public health and orthopedic diseases to address unmet medical needs. Discover more at www.ocugen.com and follow us on Twitter and LinkedIn.
Cautionary Note on Forward-Looking Statements This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995, which are subject to risks and uncertainties. We may, in some cases, use terms such as “predicts,” “believes,” “potential,” “proposed,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Such statements are subject to numerous important factors, risks, and uncertainties that may cause actual events or results to differ materially from our current expectations. These and other risks and uncertainties are more fully described in our periodic filings with the Securities and Exchange Commission (SEC), including the risk factors described in the section entitled “Risk Factors” in the quarterly and annual reports that we file with the SEC. Any forward-looking statements that we make in this press release speak only as of the date of this press release. Except as required by law, we assume no obligation to update forward-looking statements contained in this press release whether as a result of new information, future events, or otherwise, after the date of this press release.
Contact: Tiffany Hamilton Head of Communications IR@ocugen.com
Company to host conference call and webcast at 4:30 p.m. Eastern Time on Monday, November 14, 2022
NEWTOWN, Pa., Nov. 07, 2022 (GLOBE NEWSWIRE) — Onconova Therapeutics, Inc. (NASDAQ: ONTX), (“Onconova”), a clinical-stage biopharmaceutical company focused on discovering and developing novel products for patients with cancer, today announced that the Company intends to release its third quarter 2022 financial results on Monday, November 14, 2022. Management plans to host a conference call and live webcast at 4:30 p.m. ET on the same day to discuss these results and provide an update on its pipeline programs.
Conference Call and Webcast Information
Interested parties who wish to participate in the conference call may do so by dialing (800) 715-9871 for domestic and (646) 307-1963 for international callers and using conference ID 6078502.
Those interested in listening to the conference call via the internet may do so by visiting the investors and media page on the Company’s website at www.onconova.com and clicking on the webcast link. In addition to the live webcast, a replay will be available on the Onconova website for 90 days following the call.
About Onconova Therapeutics, Inc.
Onconova Therapeutics is a clinical-stage biopharmaceutical company focused on discovering and developing novel products for patients with cancer. The Company has proprietary targeted anti-cancer agents designed to disrupt specific cellular pathways that are important for cancer cell proliferation.
Onconova’s novel, proprietary multi-kinase inhibitor narazaciclib (formerly ON 123300) is being evaluated in two separate and complementary Phase 1 dose-escalation and expansion studies. These trials are currently underway in the United States and China.
Onconova’s product candidate rigosertib is being studied in an investigator-sponsored study program, including in a dose-escalation and expansion Phase 1/2a investigator-sponsored study with oral rigosertib in combination with nivolumab for patients with KRAS+ non-small cell lung cancer.
Tonix is a clinical-stage biopharmaceutical company focused on discovering, licensing, acquiring and developing therapeutics and diagnostics to treat and prevent human disease and alleviate suffering. Tonix’s portfolio is composed of immunology, rare disease, infectious disease, and central nervous system (CNS) product candidates. Tonix’s immunology portfolio includes biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-15001 which is a humanized monoclonal antibody targeting CD40-ligand being developed for the prevention of allograft and xenograft rejection and for the treatment of autoimmune diseases. A Phase 1 study of TNX-1500 is expected to be initiated in the second half of 2022. Tonix’s rare disease portfolio includes TNX-29002 for the treatment of Prader-Willi syndrome. TNX-2900 has been granted Orphan-Drug Designation by the FDA. Tonix’s infectious disease pipeline includes a vaccine in development to prevent smallpox and monkeypox called TNX-8013, next-generation vaccines to prevent COVID-19, and an antiviral to treat COVID-19. Tonix’s lead vaccine candidates for COVID-19 are TNX-1840 and TNX-18504, which are live virus vaccines based on Tonix’s recombinant pox vaccine (RPV) platform. TNX-35005 (sangivamycin, i.v. solution) is a small molecule antiviral drug to treat acute COVID-19 and is in the pre-IND stage of development. TNX-102 SL6, (cyclobenzaprine HCl sublingual tablets), is a small molecule drug being developed to treat Long COVID, a chronic post-acute COVID-19 condition. Tonix expects to initiate a Phase 2 study in Long COVID in the second quarter of 2022. The Company’s CNS portfolio includes both small molecules and biologics to treat pain, neurologic, psychiatric and addiction conditions. Tonix’s lead CNS candidate, TNX-102 SL, is in mid-Phase 3 development for the management of fibromyalgia with a new Phase 3 study launched in the second quarter of 2022. Finally, TNX-13007 is a biologic designed to treat cocaine intoxication that is expected to start a Phase 2 trial in the second quarter of 2022. TNX-1300 has been granted Breakthrough Therapy Designation by the FDA.
Robert LeBoyer, Vice President, Research Analyst, Life Sciences , Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
3Q22 Reported With Clinical Progress. Tonix reported a net loss for 3Q22 of $29.0 million or $(0.69) per share. Cash balance at the end of the quarter was $140.0 million, excluding a private placement in October that raised gross proceeds of $15 million. The company made progress in several programs during the quarter, reiterating milestones for current trials and plans for new trials in 2023.
Current Trials Have Data Milestones In 2023. The Phase 3 RESILIENT study testing TNX102 SL in fibromyalgia continues to enroll patients. A planned interim analysis is expected to be announced in 2Q23. The Phase 2 PREVAIL study in Long COVID began enrolling patients in August, with an interim analysis planned for 2Q23.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
PDS Biotech is a clinical-stage immunotherapy company developing a growing pipeline of molecularly targeted cancer and infectious disease immunotherapies based on the Company’s proprietary Versamune® and Infectimune™ T-cell activating technology platforms. Our Versamune®-based products have demonstrated the potential to overcome the limitations of current immunotherapy by inducing in vivo, large quantities of high-quality, highly potent polyfunctional tumor specific CD4+ helper and CD8+ killer T-cells. PDS Biotech has developed multiple therapies, based on combinations of Versamune® and disease-specific antigens, designed to train the immune system to better recognize diseased cells and effectively attack and destroy them. The Company’s pipeline products address various cancers including HPV16-associated cancers (anal, cervical, head and neck, penile, vaginal, vulvar) and breast, colon, lung, prostate and ovarian cancers.
Robert LeBoyer, Vice President, Research Analyst, Life Sciences , Noble Capital Markets, Inc.
Refer to the full report for the price target, fundamental analysis, and rating.
Data To Be Presented At SITC. PDS Biotechnology released two PDS0101 abstracts to be presented at the SITC (Society for Immunotherapy of Cancer) meeting later this week. Data will be from the Phase 2 IMMUNOCERV trial in cervical cancer in combination with chemotherapy and radiation and the trial known as the Triple Combination Study or NCI Study, testing PDS0101 in combination with two immunomodulating agents. We believe both studies show positive results that are consistent with our expectations.
IMMUNOCERV Trial Highlights. The Phase 2 IMMUNOCERV trial is testing PDS0101 in combination with standard-of-care chemotherapy in advanced cervical cancer. Out of 17 patients enrolled in the study, 8 had been reached the 170-day status evaluation for by PET CT. Complete responses (CR) were seen in 7 out of 8 (87.5%) of the patients. One year survival was all 8 out of 8 (100%), with 7 out of 8 disease free (87.5%). One patient who only received 3 out of 5 doses showed signs of residual disease. The study is still ongoing.
This Company Sponsored Research is provided by Noble Capital Markets, Inc., a FINRA and S.E.C. registered broker-dealer (B/D).
*Analyst certification and important disclosures included in the full report. NOTE: investment decisions should not be based upon the content of this research summary. Proper due diligence is required before making any investment decision.
Patients with high-risk, locally advanced cervical cancer on IMMUNOCERV demonstrated increased tumor-infiltrating polyfunctional CD8+ (killer) T cells, and 1-year overall survival of 100%
Data from study across several checkpoint inhibitor refractory HPV-positive cancers demonstrate an increase in HPV-specific T cells following treatment with PDS0101-based triple combination
FLORHAM PARK, N.J., Nov. 07, 2022 (GLOBE NEWSWIRE) — PDS Biotechnology Corporation (Nasdaq: PDSB), a clinical-stage immunotherapy company developing a growing pipeline of targeted immunotherapies for cancer and infectious disease, today announced upcoming poster presentations of clinical data from two Phase 2 clinical trials of PDS0101 at the 37th Annual Meeting for the Society for Immunotherapy of Cancer (SITC 2022) being held November 8-12, 2022 in Boston. PDS0101 is PDS Biotech’s lead candidate being developed as a potential treatment for HPV-positive cancers.
The first abstract accepted for presentation, titled, “IMMUNOCERV, an ongoing Phase II trial combining PDS0101, an HPV-specific T cell immunotherapy, with chemotherapy and radiation for treatment of locally advanced cervical cancers,” highlights data from The University of Texas MD Anderson Cancer Center-led IMMUNOCERV Phase 2 clinical trial (NCT04580771). The study is investigating PDS0101 in combination with standard-of-care chemoradiotherapy (CRT) for the potential treatment of cervical cancer in patients with large tumors over 5cm in size and/or cancer that has spread to the lymph nodes (lymph node metastasis). Highlights from the study being presented at SITC 2022 include:
17 patients have been enrolled in the trial.
8 of the 17 patients had completed a Day 170 post-treatment Positron Emission Tomography, Computed Tomography (PET CT) scan to assess the status of the cancer.
87.5% (7/8) of patients treated with the combination of PDS0101 and CRT demonstrated a complete response (CR) on Day 170 by PET CT. One patient who received 3 of the 5 scheduled doses of PDS0101 showed signs of residual disease.
In comparison, 74.1% (40/54) of locally advanced patients who received CRT alone and were monitored at The University of Texas MD Anderson Cancer Center on a prospective protocol independent of IMMUNOCERV had a CR on PET CT at Day 170.
The 1-year overall survival is 100% (8/8) in patients treated with the combination of PDS0101 and CRT.
The observed 1-year disease-free survival rate for IMMUNOCERV patients is 87.5% (7/8).
Patients treated with the combination of PDS0101 and CRT had a 71% increase in multi-cytokine-inducing (polyfunctional) killer (CD8+) T cells within the tumors from baseline to end of treatment (38% to 65%). This increase in activated T cells was not seen in patients receiving standard-of-care CRT.
Toxicity of PDS0101 was limited to low-grade local injection site reactions.
The second abstract, titled “Immune Correlates Associated with Clinical Benefit in Patients with Checkpoint Refractory HPV-Associated Malignancies Treated with Triple Combination Immunotherapy,” reports data from the Phase 2 triple combination trial (NCT04287868), which is being led by the Center for Cancer Research at the National Cancer Institute (NCI), part of the National Institutes of Health. The study is investigating PDS0101 in combination with two investigational immune-modulating agents: M9241, a tumor-targeting IL-12 (immunocytokine), and bintrafusp alfa, a bifunctional checkpoint inhibitor (PD-L1/ TGF-β). The triple combination is being studied in checkpoint inhibitor (CPI)-naïve and -refractory patients with advanced HPV-positive anal, cervical, head and neck, vaginal, and vulvar cancers who have failed prior therapy. For most patients who are CPI refractory, there is no effective therapy. The immune correlates before and after treatment in the CPI refractory patient population were studied. Highlights from the study being presented at SITC 2022 include:
A more than two-fold increase in HPV16-specific T cells in the blood of 79% (11/14 tested) of the evaluated patients.
Immune responses were associated with increases in natural killer cells, soluble granzyme B (associated with active killer T cells), IFN-γ, TNF-α, etc., two weeks after the first treatment cycle thus signaling a pro-inflammatory response.
These immunogenicity findings highlight the potential role of the combination in altering immune suppressive forces, and support previously announced results documenting promising clinical outcomes in the CPI-refractory population receiving the triple combination.
“We are very pleased that research describing PDS0101’s therapeutic potential will be highlighted in two poster presentations at SITC 2022, including encouraging preliminary efficacy results from the ongoing IMMUNOCERV Phase 2 clinical trial,” said Dr. Frank Bedu-Addo, CEO of PDS Biotech. “Taken together, the data being presented at SITC 2022 demonstrate the potential ability of PDS0101 to elicit in patients the right type and quality of therapeutic immune response. This seems to allow PDS0101 to work in combination with a variety of therapeutic agents to generate clinical responses that appear to exceed current standards of care and allow for improved outcomes in patients with HPV-positive cancers. We look forward to continued progression of our Phase 2 clinical trials evaluating the efficacy, safety and tolerability of PDS0101 in combination with other therapies.”
Details of the posters being presented at SITC 2022 are as follows:
Abstract Number: 674 Abstract Title: IMMUNOCERV, an ongoing Phase II trial combining PDS0101, an HPV-specific T cell immunotherapy, with chemotherapy and radiation for treatment of locally advanced cervical cancers Presenting Author: Dr. Ann Klopp, The University of Texas MD Anderson Cancer Center Session Date: Friday, Nov. 11
Abstract Number: 695 Abstract Title: Immune correlates associated with clinical benefit in patients with checkpoint refractory HPV-associated malignancies treated with triple combination immunotherapy Presenting Author: Dr. Meg Goswami, National Cancer Institute Session Date: Thursday, Nov. 10
About PDS Biotechnology PDS Biotech is a clinical-stage immunotherapy company developing a growing pipeline of targeted cancer and infectious disease immunotherapies based on our proprietary Versamune® and Infectimune™ T cell-activating technology platforms. We believe our targeted Versamune® based candidates have the potential to overcome the limitations of current immunotherapy by inducing large quantities of high-quality, potent polyfunctional tumor specific CD4+ helper and CD8+ killer T cells. To date, our lead Versamune® clinical candidate, PDS0101, has demonstrated the potential to reduce tumors and stabilize disease in combination with approved and investigational therapeutics in patients with a broad range of HPV-expressing cancers in multiple Phase 2 clinical trials. Our Infectimune™ based vaccines have also demonstrated the potential to induce not only robust and durable neutralizing antibody responses, but also powerful T cell responses, including long-lasting memory T cell responses in pre-clinical studies to date. To learn more, please visit www.pdsbiotech.com or follow us on Twitter at @PDSBiotech.
About PDS0101
PDS Biotech’s lead candidate, PDS0101, combines the utility of the Versamune® platform with targeted antigens in HPV-expressing cancers. In partnership with Merck & Co., PDS Biotech is evaluating a combination of PDS0101 and KEYTRUDA® in a Phase 2 study in first-line treatment of recurrent or metastatic head and neck cancer, and also in second line treatment of recurrent or metastatic head and neck cancer in patients who have failed prior checkpoint inhibitor therapy. A National Cancer Institute-supported Phase 2 clinical study of PDS0101 in a triple combination therapy is also being conducted in checkpoint inhibitor refractory patients with multiple advanced HPV-associated cancers. A third Phase 2 clinical trial in first line treatment of locally advanced cervical cancer is being led by The University of Texas MD Anderson Cancer Center. A final Phase 2 clinical trial of PDS0101 monotherapy in first line treatment of newly diagnosed patients HPV16+ head and neck cancer patients is being conducted at the Mayo Clinic.
KEYTRUDA® is a registered trademark of Merck Sharp and Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Forward Looking Statements This communication contains forward-looking statements (including within the meaning of Section 21E of the United States Securities Exchange Act of 1934, as amended, and Section 27A of the United States Securities Act of 1933, as amended) concerning PDS Biotechnology Corporation (the “Company”) and other matters. These statements may discuss goals, intentions and expectations as to future plans, trends, events, results of operations or financial condition, or otherwise, based on current beliefs of the Company’s management, as well as assumptions made by, and information currently available to, management. Forward-looking statements generally include statements that are predictive in nature and depend upon or refer to future events or conditions, and include words such as “may,” “will,” “should,” “would,” “expect,” “anticipate,” “plan,” “likely,” “believe,” “estimate,” “project,” “intend,” “forecast,” “guidance”, “outlook” and other similar expressions among others. Forward-looking statements are based on current beliefs and assumptions that are subject to risks and uncertainties and are not guarantees of future performance. Actual results could differ materially from those contained in any forward-looking statement as a result of various factors, including, without limitation: the Company’s ability to protect its intellectual property rights; the Company’s anticipated capital requirements, including the Company’s anticipated cash runway and the Company’s current expectations regarding its plans for future equity financings; the Company’s dependence on additional financing to fund its operations and complete the development and commercialization of its product candidates, and the risks that raising such additional capital may restrict the Company’s operations or require the Company to relinquish rights to the Company’s technologies or product candidates; the Company’s limited operating history in the Company’s current line of business, which makes it difficult to evaluate the Company’s prospects, the Company’s business plan or the likelihood of the Company’s successful implementation of such business plan; the timing for the Company or its partners to initiate the planned clinical trials for PDS0101, PDS0203 and other Versamune® and Infectimune™ based product candidates; the future success of such trials; the successful implementation of the Company’s research and development programs and collaborations, including any collaboration studies concerning PDS0101, PDS0203 and other Versamune® and Infectimune™ based product candidates and the Company’s interpretation of the results and findings of such programs and collaborations and whether such results are sufficient to support the future success of the Company’s product candidates; the success, timing and cost of the Company’s ongoing clinical trials and anticipated clinical trials for the Company’s current product candidates, including statements regarding the timing of initiation, pace of enrollment and completion of the trials (including the Company’s ability to fully fund its disclosed clinical trials, which assumes no material changes to our currently projected expenses), futility analyses, presentations at conferences and data reported in an abstract, and receipt of interim or preliminary results (including, without limitation, any preclinical results or data), which are not necessarily indicative of the final results of the Company’s ongoing clinical trials; any Company statements about its understanding of product candidates mechanisms of action and interpretation of preclinical and early clinical results from its clinical development programs and any collaboration studies; and other factors, including legislative, regulatory, political and economic developments not within the Company’s control, including unforeseen circumstances or other disruptions to normal business operations arising from or related to COVID-19. The foregoing review of important factors that could cause actual events to differ from expectations should not be construed as exhaustive and should be read in conjunction with statements that are included herein and elsewhere, including the risk factors included in the Company’s annual and periodic reports filed with the SEC. The forward-looking statements are made only as of the date of this press release and, except as required by applicable law, the Company undertakes no obligation to revise or update any forward-looking statement, or to make any other forward-looking statements, whether as a result of new information, future events or otherwise.
Versamune® is a registered trademark and Infectimune™ is a trademark of PDS Biotechnology.
Five Potentially Pivotal Phase 2 or 3 Studies for CNS Programs Expected to be in the Clinic by First Quarter 2023
Data from Planned Interim Analyses of TNX-102 SL in Phase 3 Fibromyalgia Study and Phase 2 Long COVID Study Expected Second Quarter 2023
Advanced Development Center in Dartmouth, Mass. and Infectious Disease Research and Development Facility in Frederick, Md. Operational
Cash and Cash Equivalents Totaled Approximately $140 Million at September 30, 2022
CHATHAM, N.J., Nov. 07, 2022 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP) (Tonix or the Company), a clinical-stage biopharmaceutical company, today announced financial results for the third quarter ended September 30, 2022, and provided an overview of recent operational highlights.
“Tonix continues to make meaningful progress in the development of multiple programs within its robust pipeline, having already commenced a confirmatory Phase 3 study for fibromyalgia in the second quarter of this year and a potentially pivotal Phase 2 study for Long COVID in the third quarter of this year,” said Seth Lederman, M.D., Chief Executive Officer of Tonix. “We look forward to the interim data from both of these TNX-102 SL studies in the second quarter of 2023. Additionally, we look forward to advancing our other central nervous system or CNS product candidates including TNX-102 SL for PTSD, TNX-1900 for chronic migraine, TNX-1300 for cocaine intoxication, and TNX-601 ER for depression, all of which we expect to be in the clinic by first quarter 2023. Finally, we continue to make strides in immunology with TNX-1500 for preventing organ transplant rejection expected to enter into a Phase 1 study in the first half of 2023, as well as in infectious diseases with TNX-801, a vaccine to prevent smallpox and monkeypox, expected to enter into a Phase 1 study also in the first half of next year.”
Recent Highlights—Key Product Candidates*
Central Nervous System (CNS) Pipeline
TNX-102 SL (cyclobenzaprine HCl sublingual tablet): small molecule for the management of fibromyalgia (FM)
Enrollment continues in the RESILIENT study, a double-blind, randomized, placebo-controlled, potentially pivotal confirmatory Phase 3 study of TNX-102 SL for the management of fibromyalgia. Results from a planned interim analysis are expected in the second quarter of 2023.
TNX-102 SL for the treatment of Long COVID, also known as Post-Acute Sequelae of COVID-19 (PASC)
Enrollment continues in the PREVAIL study, a potentially pivotal Phase 2 study of TNX-102 SL for Long COVID. Results from a planned interim analysis are currently anticipated in the second quarter of 2023.
Tonix presented data from a previously announced retrospective observational database study in patients with Long COVID at the International Association for the Study of Pain (IASP) 2022 World Congress on Pain. The poster presentation titled, “Retrospective Observational Database Study of Patients with Long COVID with Multi-site Pain, Fatigue, and Insomnia: A Real-World Analysis of Symptomatology and Opioid Use,” included data showing that approximately 40% of patients had fibromyalgia-like multi-site pain, the rate of opioid use in Long COVID patients with multi-site pain was 34%, which increased to approximately 50% when sleep disturbance was also present. These findings support the feasibility of the currently enrolling Phase 2 study for patients with Long COVID whose symptoms overlap with fibromyalgia.
TNX-102 SL for the treatment of Posttraumatic Stress Disorder (PTSD)
Tonix expects to begin enrolling a Phase 2 study of TNX-102 SL in police in Kenya in the fourth quarter of 2022.
Tonix expects to initiate a new, potentially pivotal, Phase 2 clinical study of TNX-1300 for the treatment of cocaine intoxication in the first quarter of 2023, pending agreement with the U.S. Food and Drug Administration (FDA).
In August 2022, Tonix received a Cooperative Agreement grant from the National Institute on Drug Abuse (NIDA), part of the National Institutes of Health (NIH), to support development of TNX-1300.
TNX-1300 has been granted Breakthrough Therapy designation by the FDA.
TNX-1900 (intranasal potentiated oxytocin): small peptide for migraine, craniofacial pain, insulin resistance and related disorders, and obesity associated binge eating disorder
The Company expects to begin enrollment in a Phase 2 study of TNX-1900 for the prevention of migraine headache in chronic migraineurs in the fourth quarter of 2022.
Tonix announced that U.S. Patent 11,389,473 issued in July 2022. The patent, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use” claims methods and compositions for treating pain, including migraine headaches, using intranasal magnesium-containing oxytocin formulations. This patent, excluding possible patent term extensions, is expected to provide Tonix with U.S. market exclusivity until January 2036.
TNX-601 ER (tianeptine hemioxalate extended-release tablets): a once-daily small molecule for the treatment of major depressive disorder (MDD), PTSD, and neurocognitive dysfunction associated with corticosteroid use.
In October 2022, Tonix announced that the FDA has cleared the Investigational New Drug (IND) application to support a Phase 2 study of TNX-601 ER for the treatment of MDD, which the Company expects to initiate in the first quarter of 2023.
TNX-601 ER is being developed as a monotherapy and first-line treatment for MDD. No tianeptine-containing product has been approved by the FDA.
Rare Disease Pipeline
TNX-2900 (intranasal potentiated oxytocin): small peptide for the treatment of Prader-Willi syndrome (PWS)
In July 2022, Tonix delivered a presentation titled, “TNX-2900 (Intranasal Oxytocin + Magnesium) in Development for the Treatment of Hyperphagia in Adolescents and Young Adults with Prader-Willi Syndrome” at the World Orphan Drug Congress USA.
TNX-2900 has been granted Orphan Drug designation from the FDA for the treatment of PWS.
Immunology Pipeline
TNX-1500 (anti-CD40L monoclonal antibody): third generation monoclonal antibody for prophylaxis of organ transplant rejection and treatment of autoimmune disorders.
Tonix announced data from three oral presentations at the 29th International Congress of The Transplantation Society (TTS 2022) by faculty at the Center for Transplantation Sciences, Massachusetts General Hospital for TNX-1500 targeting CD40-ligand (CD40L), which is also known as CD154.
The presentations titled, “Long-Term Rejection Free Renal Allograft Survival with Fc-Modified Anti-CD154 Antibody Monotherapy in Nonhuman Primates,” and “Monotherapy with TNX-1500, a Fc-Modified Anti-CD154mAb, Prolongs Cardiac Allograft Survival in Cynomolgus Monkeys,” include data demonstrating that TNX-1500 treatment showed activity in preventing organ rejection and was well tolerated in non-human primates. These presentations suggested that blockade of CD40L with TNX-1500 monotherapy consistently and safely prevented pathologic alloimmunity in non-human primate cardiac and kidney allograft models without clinical thrombosis
The presentation titled, “Long-term (>1 year) Rejection-Free Survival of Kidney Xenografts with Triple Xenoantigen Knockout and Multiple Human Transgenes in NonHuman Primates,” includes data demonstrating that TNX-1500 treatment showed activity in preventing xenograft kidney rejection and was well tolerated in non-human primates. These presentations suggested that blockade of CD40L with TNX-1500 monotherapy consistently and safely prevented pathologic xenoimmunity in non-human primate kidney xenograft models without clinical thrombosis.
A Phase 1 study of TNX-1500 is expected to start in the first half of 2023.
Infectious Disease Pipeline
TNX-801 (live horsepox virus vaccine for percutaneous administration): vaccine against smallpox and monkeypox designed as a single-administration vaccine to elicit T cell immunity
As previously mentioned, Tonix announced a collaboration with the Kenya Medical Research Institute (KEMRI) to plan, seek regulatory approval for and conduct a Phase 1 clinical study in Kenya to develop TNX-801 as a vaccine to protect against monkeypox and smallpox. The study is expected to start in the first half of 2023.
Tonix presented data from a research collaboration with The University of Alberta in a poster presentation at the 4th Symposium of the Canadian Society for Virology on June 5, 2022. The poster titled, “Synthetic Chimeric Horsepox Virus (scHPXV) Vaccination Protects Macaques from Monkeypox,” describes data from animals vaccinated with TNX-801 to protect against monkeypox. The poster presentation reports that all animals (n=8) vaccinated with TNX-801 were fully protected with sterilizing immunity from a challenge with intra-tracheal monkeypox. The vaccinations with TNX-801 were well tolerated. Synthetic horsepox virus is the basis for the Company’s TNX-801 vaccine in development to protect against monkeypox and smallpox and for the Company’s Recombinant Pox Virus (RPV) platform to protect against other pathogens, including SARS-CoV-2.
*All of Tonix’s product candidates are investigational new drugs or biologics and have not been approved for any indication.
Recent Highlights–Financial
As of September 30, 2022, Tonix had $140.0 million of cash and cash equivalents, compared to $178.7 million as of December 31, 2021.
In October 2022, Tonix issued 1,400,000 shares of Series A convertible redeemable preferred stock and 100,000 shares of Series B convertible redeemable preferred stock to certain institutional investors in a private placement for gross proceeds of $15.0 million. The Company expects to use the proceeds to redeem the preferred stock.
Cash used in operations was approximately $23.5 million for the three months ended September 30, 2022, compared to $12.9 million for the same period in 2021. The increase in cash outlays was primarily due to an increase in research and development activities. Capital expenditures were approximately $8.8 million for the three months ending September 30, 2022 compared to $7.8 million for the same period in 2021. The increase was primarily due to the continued buildout of the ADC in North Dartmouth, Mass.
Third Quarter 2022 Financial Results
Research and development (R&D) expenses for the three months ended September 30, 2022 were $22.2 million, compared to $13.1 million for the same period in 2021. The increase is predominately due to increased clinical, manufacturing, non-clinical, employee-related and laboratory expenses. The Company continues to expect R&D expenses to increase throughout the remainder of 2022 as it moves its clinical development programs forward and invests in its development pipeline.
General and administrative (G&A) expenses for the three months ended September 30, 2022 were $7.4 million, compared to $5.5 million for the same period in 2021. The increase is primarily due to increased employee-related and financial reporting expenses.
Net loss available to common stockholders was $29.0 million, or $0.69 per share, basic and diluted, for the three months ended September 30, 2022, compared to net loss of $18.5 million, or $1.60 per share, basic and diluted, for the same period in 2021. The basic and diluted weighted average common shares outstanding for the three months ended September 30, 2022 was 41,944,289 compared to 11,581,367 shares for the same period in 2021.
Tonix Pharmaceuticals Holding Corp.*
Tonix is a clinical-stage biopharmaceutical company focused on discovering, licensing, acquiring and developing therapeutics to treat and prevent human disease and alleviate suffering. Tonix’s portfolio is composed of central nervous system (CNS), rare disease, immunology and infectious disease product candidates. Tonix’s CNS portfolio includes both small molecules and biologics to treat pain, neurologic, psychiatric and addiction conditions. Tonix’s lead CNS candidate, TNX-102 SL (cyclobenzaprine HCl sublingual tablet), is in mid-Phase 3 development for the management of fibromyalgia with a new Phase 3 study launched in the second quarter of 2022 and interim data expected in the second quarter of 2023. TNX-102 SL is also being developed to treat Long COVID, a chronic post-acute-COVID-19 condition. Tonix initiated a Phase 2 study in Long COVID in the third quarter of 2022 and expects interim data in the second quarter of 2023. TNX-1300 (cocaine esterase) is a biologic designed to treat cocaine intoxication and has been granted Breakthrough Therapy designation by the FDA. A Phase 2 study of TNX-1300 is expected to be initiated in the first quarter of 2023. TNX-1900 (intranasal potentiated oxytocin), a small molecule in development for chronic migraine, is expected to enter the clinic with a Phase 2 study in the fourth quarter of 2022. TNX-601 ER (tianeptine hemioxalate extended-release tablets) is a once-daily formulation of tianeptine being developed as a potential treatment for major depressive disorder (MDD) with a Phase 2 study expected to be initiated in the first quarter of 2023. Tonix’s rare disease portfolio includes TNX-2900 (intranasal potentiated oxytocin) for the treatment of Prader-Willi syndrome. TNX-2900 has been granted Orphan Drug designation by the FDA. Tonix’s immunology portfolio includes biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft and xenograft rejection and for the treatment of autoimmune diseases. A Phase 1 study of TNX-1500 is expected to be initiated in the first half of 2023. Tonix’s infectious disease pipeline consists of a vaccine in development to prevent smallpox and monkeypox, next-generation vaccines to prevent COVID-19, and a platform to make fully human monoclonal antibodies to treat COVID-19. TNX-801, Tonix’s vaccine in development to prevent smallpox and monkeypox, also serves as the live virus vaccine platform or recombinant pox vaccine (RPV) platform for other infectious diseases. A Phase 1 study of TNX-801 is expected to be initiated in Kenya in the first half of 2023. Tonix’s lead vaccine candidate for COVID-19 is TNX-1850, a live virus vaccines based on Tonix’s recombinant pox live virus vector vaccine platform.
*All of Tonix’s product candidates are investigational new drugs or biologics and have not been approved for any indication.
This press release and further information about Tonix can be found at www.tonixpharma.com.
Forward Looking Statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; delays and uncertainties caused by the global COVID-19 pandemic; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2021, as filed with the Securities and Exchange Commission (the “SEC”) on March 14, 2022, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
TONIX PHARMACEUTICALS HOLDING CORP. CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS (In Thousands, Except Share and Per Share Amounts) (unaudited)
Three Months Ended September 30,
Nine Months Ended September 30,
2022
2021
2022
2021
COSTS AND EXPENSES:
Research and development
$
22,201
$
13,082
$
57,202
$
46,542
General and administrative
7,390
5,453
22,161
16,291
29,591
18,535
79,363
62,833
Operating loss
(29,591
)
(18,535
)
(79,363
)
(62,833
)
Interest income, net
610
7
825
99
Net loss
(28,981
)
(18,528
)
(78,538
)
(62,734
)
Preferred stock deemed dividend
—
—
4,255
—
Net loss available to common stockholders
$
(28,981
)
$
(18,528
)
$
(82,793
)
$
(62,734
)
Net loss per common share, basic and diluted
$
(0.69
)
$
(1.60
)
$
(3.06
)
$
(6.02
)
Weighted average common shares outstanding, basic and diluted
1The condensed consolidated balance sheet for the year ended December 31, 2021 has been derived from the audited financial statements but do not include all of the information and footnotes required by accounting principles generally accepted in the United States for complete financial statements.
What is Inflammation? Two Immunologists Explain How the Body Responds to Everything from Stings to Vaccination and Why it Sometimes Goes Wrong
When your body fights off an infection, you develop a fever. If you have arthritis, your joints will hurt. If a bee stings your hand, your hand will swell up and become stiff. These are all manifestations of inflammation occurring in the body.
We are two immunologists who study how the immune system reacts during infections, vaccination and autoimmune diseases where the body starts attacking itself.
This article was republished with permission from The Conversation, a news site dedicated to sharing ideas from academic experts. It represents the research-based findings and thoughts of Prakash Nagarkatti, Professor of Pathology, Microbiology and Immunology, University of South Carolina and Mitzi Nagarkatti Professor of Pathology, Microbiology and Immunology, University of South Carolina
While inflammation is commonly associated with the pain of an injury or the many diseases it can cause, it is an important part of the normal immune response. The problems arise when this normally helpful function overreacts or overstays its welcome.
What is Inflammation?
Generally speaking, the term inflammation refers to all activities of the immune system that occur where the body is trying to fight off potential or real infections, clear toxic molecules or recover from physical injury. There are five classic physical signs of acute inflammation: heat, pain, redness, swelling and loss of function. Low-grade inflammation might not even produce noticeable symptoms, but the underlying cellular process is the same.
Take a bee sting, for example. The immune system is like a military unit with a wide range of tools in its arsenal. After sensing the toxins, bacteria and physical damage from the sting, the immune system deploys various types of immune cells to the site of the sting. These include T cells, B cells, macrophages and neutrophils, among other cells.
The B cells produce antibodies. Those antibodies can kill any bacteria in the wound and neutralize toxins from the sting. Macrophages and neutrophils engulf bacteria and destroy them. T cells don’t produce antibodies, but kill any virus-infected cell to prevent viral spread.
Additionally, these immune cells produce hundreds of types of molecules called cytokines – otherwise known as mediators – that help fight threats and repair harm to the body. But just like in a military attack, inflammation comes with collateral damage.
The mediators that help kill bacteria also kill some healthy cells. Other similar mediating molecules cause blood vessels to leak, leading to accumulation of fluid and influx of more immune cells.
This collateral damage is the reason you develop swelling, redness and pain around a bee sting or after getting a flu shot. Once the immune system clears an infection or foreign invader – whether the toxin in a bee sting or a chemical from the environment – different parts of the inflammatory response take over and help repair the damaged tissue.
After a few days, your body will neutralize the poison from the sting, eliminate any bacteria that got inside and heal any tissue that was harmed.
Asthma is caused by inflammation that leads to swelling and a narrowing of airways in the lungs, as seen in the right cutaway in this image. BruceBlaus/Wikimedia Commons, CC BY-SA
Inflammation as a Cause of Disease
Inflammation is a double-edged sword. It is critical for fighting infections and repairing damaged tissue, but when inflammation occurs for the wrong reasons or becomes chronic, the damage it causes can be harmful.
Allergies, for example, develop when the immune system mistakenly recognizes innocuous substances – like peanuts or pollen – as dangerous. The harm can be minor, like itchy skin, or dangerous if someone’s throat closes up.
Chronic inflammation damages tissues over time and can lead to many noninfectious clinical disorders, including cardiovascular diseases, neurodegenerative disorders, obesity, diabetes and some types of cancers.
The immune system can sometimes mistake one’s own organs and tissues for invaders, leading to inflammation throughout the body or in specific areas. This self-targeted inflammation is what causes the symptoms of autoimmune diseases such as lupus and arthritis.
Another cause of chronic inflammation that researchers like us are currently studying is defects in the mechanisms that curtail inflammation after the body clears an infection.
While inflammation mostly plays out at a cellular level in the body, it is far from a simple mechanism that happens in isolation. Stress, diet and nutrition, as well as genetic and environmental factors, have all been shown to regulate inflammation in some way.
There is still a lot to be learned about what leads to harmful forms of inflammation, but a healthy diet and avoiding stress can go a long way toward helping maintain the delicate balance between a strong immune response and harmful chronic inflammation.
CARLSBAD, Calif.–(BUSINESS WIRE)–Nov. 3, 2022– Lineage Cell Therapeutics, Inc. (NYSE American and TASE: LCTX), a clinical-stage biotechnology company developing allogeneic cell therapies for unmet medical needs, today announced that it will report its third quarter 2022 financial and operating results on Thursday, November 10, 2022, following the close of the U.S. financial markets. Lineage management will also host a conference call and webcast on Thursday, November 10, 2022, at 4:30 p.m. Eastern Time/1:30 p.m. Pacific Time to discuss its third quarter 2022 financial and operating results and to provide a business update.
Interested parties may access the conference call by dialing (800) 715-9871 from the U.S. and Canada and should request the “Lineage Cell Therapeutics Call” or provide conference ID number5262180. A live webcast of the conference call will be available online in the Investors section of Lineage’s website. A replay of the webcast will be available on Lineage’s website for 30 days and a telephone replay will be available through November 17, 2022, by dialing (800) 770-2030 from the U.S. and Canada and entering conference ID number 5262180.
About Lineage Cell Therapeutics, Inc.
Lineage Cell Therapeutics is a clinical-stage biotechnology company developing novel cell therapies for unmet medical needs. Lineage’s programs are based on its robust proprietary cell-based therapy platform and associated in-house development and manufacturing capabilities. With this platform Lineage develops and manufactures specialized, terminally differentiated human cells from its pluripotent and progenitor cell starting materials. These differentiated cells are developed to either replace or support cells that are dysfunctional or absent due to degenerative disease or traumatic injury or administered as a means of helping the body mount an effective immune response to cancer. Lineage’s clinical and preclinical programs are in markets with billion dollar opportunities and include five allogeneic (“off-the-shelf”) product candidates: (i) OpRegen, a retinal pigment epithelial cell therapy in development for the treatment of geographic atrophy secondary to age-related macular degeneration, is being developed under a worldwide collaboration with Roche and Genentech, a member of the Roche Group; (ii) OPC1, an oligodendrocyte progenitor cell therapy in Phase 1/2a development for the treatment of acute spinal cord injuries; (iii) VAC2, a dendritic cell therapy produced from Lineage’s VAC technology platform for immuno-oncology and infectious disease, currently in Phase 1 clinical development for the treatment of non-small cell lung cancer; (iv) ANP1, an auditory neuronal progenitor cell therapy for the potential treatment of auditory neuropathy; and (v) PNC1, a photoreceptor neural cell therapy for the potential treatment of vision loss due to photoreceptor dysfunction or damage. For more information, please visit www.lineagecell.com or follow the company on Twitter @LineageCell.