RICHMOND, Va.–(BUSINESS WIRE)– Lucky Strike Entertainment (NYSE: LUCK), one of the world’s premier Owner/ Operators of location-based entertainment, announced today the appointment of two esteemed executives to its Board of Directors: Richard Born, a pioneering force in hospitality-focused real estate, and Jason Harinstein, a recognized leader in finance, technology, and business strategy. Their appointments are effective June 23, 2025.
These strategic additions bring a wealth of expertise in hospitality, real estate, finance, and technology to Lucky Strike’s board, reinforcing the company’s commitment to innovation, growth, and world-class guest experiences. Mr. Born will serve on the Nominating and Corporate Governance Committee of the Board, and Mr. Harinstein will join the Board’s Audit and Compensation Committees.
Mr. Born is widely regarded as one of the most influential developers in New York’s boutique hotel landscape. As co-founder of BD Hotels, he has spent over 35 years transforming properties into cultural and hospitality landmarks. His portfolio includes interests in more than 25 hotels and 20 additional real estate holdings, and he remains actively involved in the management and operations of over half of these assets. His projects include iconic properties such as the Hotel Chelsea, the Bowery Hotel, the Mercer, and the Ludlow, which have redefined the guest experience and helped shape the hospitality scene in New York as well as other major markets. Mr. Born holds a medical doctorate from the New York University School of Medicine.
Mr. Harinstein brings over two decades of executive leadership from some of the industry’s most innovative and data-driven companies. He currently serves as Chief Financial Officer of Collectors Holdings Inc., a premier platform for authentication and digital marketplaces. Prior to that, he was the Chief Financial Officer of Flatiron Health, a healthcare technology company, and held senior roles at Google and Groupon, where he helped drive corporate strategy, and business development during key phases of expansion. Mr. Harinstein is also a member of the board of directors of Groupon and Funko. He holds a master’s of business administration from the University of Chicago Booth School of Business.
“Richard and Jason are best-in-class leaders in their respective fields—each bringing a unique blend of operational insight, visionary thinking, and financial discipline,” said Thomas Shannon, Founder, Chairman, and CEO of Lucky Strike Entertainment. “Their addition to our board reflects our commitment to assembling world-class talent to guide our continued growth, innovation, and long-term vision.”
The appointments underscore Lucky Strike’s continued investment in top-tier leadership as the company continues to expand its footprint and deepen its position as an industry leader.
About Lucky Strike Entertainment
Lucky Strike Entertainment is one of the world’s premier location-based entertainment platforms. With over 360 locations across North America, Lucky Strike Entertainment provides experiential offerings in bowling, amusements, water parks, and family entertainment centers. The company also owns the Professional Bowlers Association, the major league of bowling and a growing media property that boasts millions of fans around the globe. For more information on Lucky Strike Entertainment, please visit IR.LuckyStrikeEnt.com.
Proposed reverse merger with OrthoCellix, a wholly-owned subsidiary of Ocugen, to create Nasdaq-listed, late clinical-stage regenerative cell therapy company with a first-in-class technology platform, focused on orthopedic diseases
OrthoCellix is developing the Phase 3-ready NeoCart® as an autologous cartilage implant technology utilizing patient cells to repair articular cartilage defects of the knee
PHILADELPHIA and MALVERN, Pa., June 23, 2025 (GLOBE NEWSWIRE) — Carisma Therapeutics Inc. (Nasdaq: CARM) (Carisma) and OrthoCellix, Inc. (OrthoCellix), a wholly-owned subsidiary of Ocugen, Inc. (Nasdaq: OCGN) (Ocugen), a clinical-stage company developing regenerative cell therapies for orthopedic diseases, today jointly announced that they have entered into a definitive merger agreement to combine the companies in an all-stock transaction. The combined company will focus on the development of OrthoCellix’s NeoCart® technology for the treatment of knee articular cartilage defects and plans to initiate a U.S. Food and Drug Administration (FDA)-endorsed Phase 3 clinical trial for NeoCart®.
“We believe merging OrthoCellix with Carisma will allow us to create a publicly-traded company focused on the development of NeoCart® and provide value for both Ocugen and Carisma stockholders while unlocking true market potential of NeoCart®,” said Dr. Shankar Musunuri, Chairman, Chief Executive Officer, and Co-founder of Ocugen. “We believe NeoCart® has tremendous potential to deliver a truly transformative approach to cartilage repair, and we’ve established OrthoCellix with dedicated resources to bring this revolutionary technology to the patients who desperately need it.”
“Carisma evaluated a range of strategic alternatives, and we believe this proposed transaction represents an opportunity to deliver significant value to our stockholders,” said Steven Kelly, President and Chief Executive Officer, of Carisma. “OrthoCellix is strongly positioned with its NeoCart® platform, a dedication to developing regenerative cell therapies, and a well-credentialed management team to lead the combined company.”
About OrthoCellix’s NeoCart® Portfolio
OrthoCellix is developing NeoCart® as an autologous cartilage implant technology utilizing patient cells to repair articular cartilage defects of the knee. The novel platform merges a fortified 3D scaffold and patented bioprocessing technology to grow chondrocytes—the cells responsible for maintaining cartilage health—to produce adolescent-like cartilage at the time of implant. NeoCart® has the potential to accelerate healing and reduce pain by creating a similar, functional joint surface to help patients return to normal activities and prevent complications associated with articular cartilage damage.
OrthoCellix anticipates launching its Phase 3 clinical trial by the end of 2025. Previously, NeoCart® received Regenerative Medicine Advanced Therapy (RMAT) designation and concurrence from the FDA on a single, confirmatory Phase 3 clinical trial to enable submission of a Biologics License Application.
About the Proposed Transactions
Under the terms of the merger agreement, OrthoCellix will merge with and into a wholly-owned subsidiary of Carisma, with OrthoCellix continuing as a wholly-owned subsidiary of Carisma and the surviving company of the Merger. Carisma will issue to the pre-merger OrthoCellix stockholder shares of Carisma common stock as merger consideration in exchange for the cancellation of shares of capital stock of OrthoCellix. Carisma also expects to enter into subscription agreements for a private financing with Ocugen and other select investors, which is expected to close concurrently with the completion of the merger, to enable the combined company to complete the Phase 3 trial of NeoCart® without any additional cost or investment from Ocugen. In connection with the closing of the proposed transactions, Carisma stockholders will be issued contingent value rights representing the right to receive certain payments from proceeds received by the combined company, if any, related to Carisma’s pre-transaction legacy assets.
Under the terms of the merger agreement, upon the closing of the proposed transactions and after giving effect to the contemplated $25.0 million concurrent financing, OrthoCellix’s stockholder and the other participants in the concurrent financing are expected to own approximately 90% of the combined company, and existing Carisma stockholders are expected to own approximately 10% of the combined company, each on a fully diluted basis. The percentage of the combined company that each company’s former stockholders will own after completion of the merger is subject to adjustment based on Carisma’s net cash at the closing and the proceeds from the concurrent financing, among other adjustments, in each case as described in the merger agreement.
Upon the closing of the proposed transactions, “Carisma Therapeutics Inc.” is expected to be renamed “OrthoCellix, Inc.” and trade on the Nasdaq Capital Market under the ticker symbol ‘OCLX.’
The transaction has been unanimously approved by the board of directors of both companies and is expected to close in the second half of 2025, subject to customary closing conditions, including approvals by the stockholders of each company and the effectiveness of a registration statement to be filed with the Securities and Exchange Commission (the “SEC”) to register the shares of Carisma common stock to be issued in connection with the merger. In connection with the companies’ entry into the merger agreement, directors and officers of Carisma and OrthoCellix’s stockholder have executed support agreements, pursuant to which they have agreed to vote all of their shares of capital stock in favor of the merger or the issuance of Carisma equity in the merger, as applicable.
Advisors
Wilmer Cutler Pickering Hale and Dorr LLP is serving as legal counsel to Carisma and Lucid Capital Markets, LLC is providing a fairness opinion to Carisma’s board of directors. Chardan Capital Markets LLC is serving as M&A advisor and co-placement agent to OrthoCellix and Ocugen. Lake Street Capital Markets, LLC is co-placement agent to OrthoCellix, as a subsidiary of Ocugen, Goodwin Procter LLP is serving as legal counsel to Ocugen and OrthoCellix, and Paul Hastings LLP is serving as legal counsel to the placement agents.
About OrthoCellix
OrthoCellix is a regenerative cell therapy company dedicated to developing a first-in-class technology platform focused on cartilage defects and other orthopedic diseases to address considerable unmet medical needs. The lead program within OrthoCellix is NeoCart® with revolutionary 3D cell therapy technology designed to repair and restore articular cartilage defects in the knee. The Company has a pipeline of additional treatments based on its proprietary scaffold bioreactor and adhesive. OrthoCellix will utilize the Good Manufacturing Practice facility established by Ocugen to support OrthoCellix’s initial development of NeoCart®.
About Carisma Therapeutics
Carisma Therapeutics is a biotechnology company pioneering macrophage engineering to develop groundbreaking therapies for fibrosis and cancer. With a strong commitment to patient-centric innovation, Carisma aims to deliver scalable, next-generation solutions that transform treatment paradigms. Carisma is headquartered in Philadelphia, PA. For more information, please visit www.Carismatx.com.
Cautionary Note on Forward- Looking Statements
Certain statements in this communication, other than purely historical information, may constitute “forward-looking statements” within the meaning of the federal securities laws, including for purposes of the “safe harbor” provisions under the Private Securities Litigation Reform Act of 1995, concerning Carisma, OrthoCellix, the proposed financing and the proposed merger between Carisma and OrthoCellix (collectively, the “Proposed Transactions”) and other matters. These forward-looking statements include, but are not limited to, express or implied statements relating to Carisma’s and OrthoCellix’s management teams’ expectations, hopes, beliefs, intentions or strategies regarding the future including, without limitation, statements regarding: the structure, timing and completion of the proposed merger by and between Carisma and OrthoCellix; the Proposed Transactions and the expected effects, perceived benefits or opportunities of the Proposed Transactions; the combined company’s listing on Nasdaq after the closing of the Proposed Transactions; expectations regarding the structure, timing and completion of a concurrent financing, including investment amounts from investors, timing of closing of the Proposed Transactions, expected proceeds, expectations regarding the use of proceeds, and impact on ownership structure; the anticipated timing of the closing; the expected executive officers and directors of the combined company; each company’s and the combined company’s expected cash position at the closing and cash runway of the combined company following the proposed merger and any private financing; the future operations of the combined company, including research and development activities; the nature, strategy and focus of the combined company; the development and commercial potential and potential benefits of any product candidates of the combined company, including expectations around market exclusivity and intellectual property protection; anticipated clinical drug development activities and related timelines, including the expected timing for announcement of data and other clinical results; expectations regarding or plans for discovery, preclinical studies, clinical trials and research and development programs, in particular with respect to NeoCart®, and any developments or results in connection therewith, including the target product profile of NeoCart®; the anticipated timing of the commencement of and results from those studies and trials; the sufficiency of post-transaction resources to support the advancement of OrthoCellix’s pipeline through certain milestones and the time period over which OrthoCellix’s post-transaction capital resources will be sufficient to fund its anticipated operations; the cash balance of the combined entity at closing; expectations related to the anticipated timing of the closing of the Proposed Transactions (the “Closing”); the expectations regarding the ownership structure of the combined company; the expected trading of the combined company’s stock on Nasdaq under the ticker symbol “OCLX” after the Closing; and other statements that are not historical fact. All statements other than statements of historical fact contained in this communication are forward-looking statements. In addition, any statements that refer to projections, forecasts or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking statements. The words “opportunity,” “potential,” “milestones,” “pipeline,” “can,” “goal,” “strategy,” “target,” “anticipate,” “achieve,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intends,” “may,” “plan,” “possible,” “project,” “should,” “will,” “would” and similar expressions (including the negatives of these terms or variations of them) may identify forward-looking statements, but the absence of these words does not mean that a statement is not forward-looking. These forward-looking statements are made based on current expectations, estimates, forecasts, and projections, as well as the beliefs and assumptions of management, concerning future developments and their potential effects. There can be no assurance that future developments affecting Carisma, OrthoCellix, or the Proposed Transactions will be those that have been anticipated.
These forward-looking statements involve a number of risks and uncertainties, some of which are beyond Carisma’s or OrthoCellix’s control, or other assumptions that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. These risks and uncertainties include, but are not limited to, the risk that the conditions to the Closing or consummation of the Proposed Transactions are not satisfied, including the failure to timely obtain approval of the proposed reverse stock split from Carisma’s stockholders and the proposed merger from both Carisma’s and OrthoCellix’s stockholders, if at all; the risk that the proposed concurrent financing is not completed in a timely manner, if at all; uncertainties as to the timing of the consummation of the Proposed Transactions and the ability of each of Carisma and OrthoCellix to consummate the Proposed Transactions; risks related to Carisma’s continued listing on Nasdaq until closing of the Proposed Transactions and the combined company’s ability to remain listed following the Closing; risks related to Carisma’s and OrthoCellix’s ability to correctly estimate their respective operating expenses and their respective expenses associated with the Proposed Transactions, as applicable, pending the Closing, as well as uncertainties regarding the impact any delay in the Closing would have on the anticipated cash resources of the combined company, and other events and unanticipated spending and costs that could reduce the combined company’s cash resources; risks related to the failure or delay in obtaining required approvals from any governmental or quasi-governmental entity necessary to consummate the Proposed Transactions; the occurrence of any event, change or other circumstance or condition that could give rise to the termination of the merger agreement; the effect of the announcement or pendency of the merger on Carisma’s or OrthoCellix’s business relationships, operating results and business generally; costs related to the merger; the risk that as a result of adjustments to the exchange ratio, OrthoCellix stockholders and Carisma stockholders could own more or less of the combined company than is currently anticipated; risks related to the market price of Carisma’s common stock relative to the value suggested by the exchange ratio; the uncertainties associated with OrthoCellix’s NeoCart® portfolio, as well as risks associated with the clinical development and regulatory approval of product candidates, including potential delays in the completion of clinical trials; risks related to the inability of the combined company to obtain sufficient additional capital to continue to advance these product candidates; uncertainties in obtaining successful clinical results for product candidates and unexpected costs that may result therefrom; risks related to the failure to realize any value from product candidates being developed and anticipated to be developed in light of inherent risks and difficulties involved in successfully bringing product candidates to market; the outcome of any legal proceedings that may be instituted against Carisma, OrthoCellix or any of their respective directors or officers related to the Proposed Transactions; the ability of Carisma and OrthoCellix to obtain, maintain, and protect their respective intellectual property rights; competitive responses to the Proposed Transactions; costs of the Proposed Transactions and unexpected costs, charges or expenses resulting from the Proposed Transactions; potential adverse reactions or changes to business relationships, operating results, and business generally, resulting from the announcement or completion of the Proposed Transactions; changes in regulatory requirements and government incentives; risks associated with the possible failure to realize, or that it may take longer to realize than expected, certain anticipated benefits of the Proposed Transactions, including with respect to future financial and operating results, legislative, regulatory, political and economic developments, and those uncertainties and factors; and the risk of involvement in litigation, including securities class action litigation, that could divert the attention of the management of Carisma or the combined company, harm the combined company’s business and may not be sufficient for insurance coverage to cover all costs and damages, among others. Actual results and the timing of events could differ materially from those anticipated in such forward-looking statements as a result of these risks and uncertainties. These and other risks and uncertainties are more fully described in periodic filings with the SEC, including the factors described in the section titled “Risk Factors” in Carisma’s Annual Report on Form 10-K for the year ended December 31, 2024, which was originally filed with the SEC on March 31, 2025, as amended by Amendment No. 1 to the Annual Report on Form 10-K/A, which was filed with the SEC on April 29, 2025, subsequent Quarterly Reports on Form 10-Q filed with the SEC, and in other filings that Carisma makes and will make with the SEC in connection with the Proposed Transactions, including the Form S-4 and Proxy Statement described below under “Additional Information and Where to Find It”, as well as discussions of potential risks, uncertainties, and other important factors included in other filings by Carisma from time to time, any risk factors related to Carisma or OrthoCellix made available to you in connection with the Proposed Transactions, as well as risk factors associated with companies, such as OrthoCellix, that operate in the biopharma industry. Should one or more of these risks or uncertainties materialize, or should any of Carisma’s or OrthoCellix’s assumptions prove incorrect, actual results may vary in material respects from those projected in these forward-looking statements. Nothing in this communication should be regarded as a representation by any person that the forward-looking statements set forth herein will be achieved or that any of the contemplated results of such forward-looking statements will be achieved. You should not place undue reliance on forward-looking statements in this communication, which speak only as of the date they are made and are qualified in their entirety by reference to the cautionary statements herein. Neither Carisma nor OrthoCellix undertakes or accepts any duty to release publicly any updates or revisions to any forward-looking statements contained herein to reflect any change in its expectations with regard thereto or any change in events, conditions or circumstances on which any such statements are based, except as required by law. This communication does not purport to summarize all of the conditions, risks and other attributes of an investment in Carisma or OrthoCellix.
No Offer or Solicitation
This communication and the information contained herein is not intended to and does not constitute (i) a solicitation of a proxy, consent or approval with respect to any securities or in respect of the Proposed Transactions or (ii) an offer to sell or the solicitation of an offer to subscribe for or buy or an invitation to purchase or subscribe for any securities pursuant to the Proposed Transactions or otherwise, nor shall there be any sale, issuance or transfer of securities in any jurisdiction in contravention of applicable law. No offering of securities shall be made except by means of a prospectus meeting the requirements of Section 10 of the Securities Act of 1933, as amended, and otherwise in accordance with applicable law, or an exemption therefrom. Subject to certain exceptions to be approved by the relevant regulators or certain facts to be ascertained, the public offer will not be made directly or indirectly, in or into any jurisdiction where to do so would constitute a violation of the laws of such jurisdiction, or by use of the mails or by any means or instrumentality (including without limitation, facsimile transmission, telephone and the internet) of interstate or foreign commerce, or any facility of a national securities exchange, of any such jurisdiction.
NEITHER THE SEC NOR ANY STATE SECURITIES COMMISSION HAS APPROVED OR DISAPPROVED OF THE SECURITIES OR DETERMINED IF THIS COMMUNICATION IS TRUTHFUL OR COMPLETE.
Important Additional Information about the Proposed Transactions Will be Filed with the SEC
This communication relates to the proposed merger involving Carisma and OrthoCellix and may be deemed to be solicitation material in respect of the proposed merger. In connection with the Proposed Transactions, Carisma intends to file relevant materials with the SEC, including a registration statement on Form S-4 (the “Form S-4”) that will contain a proxy statement (the “Proxy Statement”) and prospectus. This communication is not a substitute for the Form S-4, the Proxy Statement or for any other document that Carisma may file with the SEC and/or send to Carisma’s stockholders in connection with the proposed merger. CARISMA URGES, BEFORE MAKING ANY VOTING DECISION, INVESTORS AND STOCKHOLDERS TO READ THE FORM S-4, THE PROXY STATEMENT AND ANY OTHER RELEVANT DOCUMENTS THAT MAY BE FILED WITH THE SEC, AS WELL AS ANY AMENDMENTS OR SUPPLEMENTS TO THESE DOCUMENTS, CAREFULLY AND IN THEIR ENTIRETY IF AND WHEN THEY BECOME AVAILABLE BECAUSE THEY WILL CONTAIN IMPORTANT INFORMATION ABOUT CARISMA, ORTHOCELLIX, THE PROPOSED TRANSACTIONS AND RELATED MATTERS.
Investors and stockholders will be able to obtain free copies of the Form S-4, the Proxy Statement and other documents filed by Carisma with the SEC (when they become available) through the website maintained by the SEC at www.sec.gov. Copies of the documents filed by Carisma with the SEC will also be available free of charge on Carisma’s website at www.carismatx.com, or by contacting Carisma’s Investor Relations at investors@Carismatx.com. In addition, investors and stockholders should note that Carisma communicates with investors and the public using its website at https://ir.Carismatx.com/.
Participants in the Solicitation
Carisma, OrthoCellix, and their respective directors and certain of their executive officers and other members of management may be deemed to be participants in the solicitation of proxies from Carisma’s stockholders in connection with the Proposed Transactions under the rules of the SEC. Information about Carisma’s directors and executive officers, including a description of their interests in Carisma, is included in Carisma’s most recent Annual Report on Form 10-K for the year ended December 31, 2024, which was filed with the SEC on March 31, 2025, as amended by Amendment No. 1 to the Annual Report on Form 10-K, which was filed with the SEC on April 29, 2025. Additional information regarding the persons who may be deemed participants in the proxy solicitations, including about the directors and executive officers of OrthoCellix, and a description of their direct and indirect interests, by security holdings or otherwise, will also be included in the Form S-4, the Proxy Statement and other relevant materials to be filed with the SEC when they become available. These documents can be obtained free of charge from the sources indicated above.
Vancouver, British Columbia–(Newsfile Corp. – June 20, 2025) – Nicola Mining Inc. (TSX: NIM) (OTCQB: HUSIF) (FSE: HLIA) (the “Company” or “Nicola“) is pleased to announce commencement of the 2025 Exploration Diamond Drilling Program (the “2025 Program“) at its New Craigmont Copper Project (“New Craigmont”), near Merritt, BC.
Exploration Target Generation Activities
Five priority exploration targets (Figure 1), three of which are included in Nicola’s 2025 program, have been identified through collaboration in 2025 with ALS Geoanalytics (GoldSpot Discoveries Ltd.; “GoldSpot”) using AI-based methods to analyze and correlate geophysical and geochemical data from Nicola’s large exploration database.
Figure 1. Locations of the top exploration targets based on analysis of geophysical and geochemical data in 2025 by Goldspot.
Target A corresponds well with the West Craigmont/WP target area where several holes were drilled in 2024 (see August 30, 2024 News Release). Drilling in 2024 revealed favourable alteration with follow-up potential.
Target B is a new, undrilled target identified by the Goldspot analysis.
Target C corresponds with the MARB/CAS target area where positive results from the 2024 Exploration Program made it a high priority target.
Target D corresponds with the important Titan Queen MINFILE showing. Historic and subsequent drilling and mapping in 2016 support more drilling.
Target E is another new target from the Goldspot analysis that is added to the 2025 Program.
Nicola continues to work with GoldSpot to refine the 2025 Exploration Program that will include collection of X-ray fluorescence (pXRF) and short-wave infrared (SWIR) data under the guidance of GoldSpot to ensure consistent, high-quality data acquisition aligned with New Craigmont’s exploration goals. This new data will contribute to the development of exploration targets and improve understanding of skarn and porphyry-style mineralization.
Diamond Drilling Plans
Exploration plans for the 2025 Program include 4,000-5,000 metres of diamond drilling at the MARB/CAS, West Craigmont/WP, and two new target areas generated by ALS GoldSpot (Fig. 2). The purpose of the 2025 Program is to collect geological data for target development for a potential porphyry copper system at New Craigmont. Drill core will provide valuable information on lithology, structure, alteration and mineralization, and multi-element analysis.
Figure 2. Target areas for 2025 drilling Program at the New Craigmont Project.
Drilling at MARB will follow-up near-surface porphyry-style copper-mineralization with holes designed to test a vertical mineralization trend at depth. Near surface skarn at CAS discovered in 2024 is characteristic of mineralization observed in the Embayment Zone. Additional drilling in the 2025 Program will investigate potential continuity along trend between MARB, CAS and the Embayment Zone.
Draken is a high-priority, undrilled target consisting of a cluster of copper showings discovered from Nicola’s field program in 2023 (Figure 2). Outcrops of Guichon Border Phase quartz diorite contain porphyry style quartz-feldspar veinlets with weak copper oxide minerals. Exposures at Draken exhibit some of the best-developed porphyry-style alteration documented on the New Craigmont property, and the target also coincides with high resistivity and high chargeability geophysical response.
Figure 3. Outcrop at the newly discovered Draken showing displaying several sets of quartz-K Feldspar +/- epidote veins with some of the veins containing copper oxide (inset), all of which are common features of a porphyry system.
Nicola’s objective for 2025 is to continue to target for porphyry systems by conducting the following:
Acquire an enhanced suite of geochemical data for more targeting studies with GoldSpot
Expand the extent of mineralization observed at the MARB and CAS targets
Test two new targets at West Craigmont, including Draken
Test two new targets generated by GoldSpot in the centre of the property
The estimated budget for the 2025 Program is $1.5-2 M. Nicola anticipates drilling to conclude sometime in September.
The Company will provide a separate news release on exploration at its high-grade silver Treasure Mountain Project.
Qualified Person
The scientific and technical disclosure included in this news release have been reviewed and approved by Will Whitty, P.Geo., who is the Qualified Person as defined by NI 43-101. Mr. Whitty is Vice President, Exploration for the Company.
About Nicola Mining
Nicola Mining Inc. is a junior mining company listed on the TSX-V Exchange and Frankfurt Exchange that maintains a 100% owned mill and tailings facility, located near Merritt, British Columbia. It has signed Mining and Milling Profit Share Agreements with high-grade BC-based gold projects. Nicola’s fully permitted mill can process both gold and silver mill feed via gravity and flotation processes.
The Company owns 100% of the New Craigmont Project, a property that hosts historic high-grade copper mineralization and covers an area of over 10,800 hectares along the southern end of the Guichon Batholith and is adjacent to Highland Valley Copper, Canada’s largest copper mine. The Company also owns 100% of the Treasure Mountain Property, which includes 30 mineral claims and a mineral lease, spanning an area exceeding 2,200 hectares.
Neither the TSX Venture Exchange nor its Regulation Services Provider (as that term is defined in the policies of the TSX Venture Exchange) accepts responsibility for the adequacy or accuracy of this release.
Nutriband and Kindeva have completed commercial manufacturing process scale-up for its lead product Aversa™ Fentanyl, an abuse-deterrent fentanyl patch
Nutriband is partnering with Kindeva to develop Aversa™ Fentanyl which combines Nutriband’s Aversa™ abuse-deterrent technology with Kindeva’s FDA-approved fentanyl patch
ORLANDO, Fla., June 18, 2025 (GLOBE NEWSWIRE) — Nutriband Inc. (NASDAQ:NTRB)(NASDAQ:NTRBW), a company engaged in the development of prescription transdermal pharmaceutical products, today announced that it has completed commercial manufacturing process scale-up for its lead product, Aversa™ Fentanyl, with Kindeva, a leading global contract development and manufacturing organization (CDMO) focused on drug-device combination products.
Nutriband is partnering with Kindeva to develop Aversa™ Fentanyl which combines Nutriband’s Aversa™ abuse-deterrent technology with Kindeva’s FDA-approved fentanyl patch. Aversa Fentanyl is manufactured at Kindeva’s state-of-the-art transdermal manufacturing facility located in the United States. The next step is to manufacture clinical supplies and file an Investigational New Drug (IND) application with the FDA to initiate a human abuse liability clinical study.
“We are excited to achieve this commercial development milestone with our partner, Kindeva. Completing the commercial manufacturing scale-up is an important step towards development of a commercially viable product and eventual NDA filing. This achievement demonstrates the compatibility of the Aversa™ abuse deterrent platform technology with established transdermal patch manufacturing processes. Aversa Fentanyl has the potential to be the first abuse deterrent pain patch on the market,” said Gareth Sheridan, CEO, Nutriband.
Nutriband’s AVERSA™ abuse-deterrent technology can be utilized to incorporate aversive agents into transdermal patches to prevent the abuse, diversion, misuse, and accidental exposure of drugs with abuse potential including opioids and stimulants. The AVERSA™ abuse-deterrent technology has the potential to improve the safety profile of transdermal drugs susceptible to abuse, such as fentanyl, while making sure that these drugs remain accessible to those patients who really need them.
AVERSA Fentanyl has the potential to be the world’s first abuse-deterrent opioid patch designed to deter the abuse and misuse and reduce the risk of accidental exposure of transdermal fentanyl patches. AVERSA Fentanyl has the potential to reach peak annual US sales of $80 million to $200 million.1 While initially concentrating on the US market, the unmet medical need for adequate pain management is a global problem, and our goal is to make AVERSA a global solution strategically targeting all major medical markets in the world.
The AVERSA™ abuse deterrent technology is protected by a broad international intellectual property portfolio with patents issued in 46 countries including the United States, Europe, Japan, Korea, Russia, China, Canada, Mexico, and Australia.
1 Health Advances Aversa Fentanyl market analysis report 2022
About AVERSA™ Abuse-Deterrent Transdermal Technology
Nutriband’s AVERSA™ abuse-deterrent transdermal technology incorporates aversive agents into transdermal patches to prevent the abuse, diversion, misuse, and accidental exposure of drugs with abuse potential. The AVERSA™ abuse-deterrent technology has the potential to improve the safety profile of transdermal drugs susceptible to abuse, such as fentanyl, while making sure that these drugs remain accessible to those patients who really need them. The technology is covered by a broad intellectual property portfolio with patents granted in the United States, Europe, Japan, Korea, Russia, China, Canada, Mexico, and Australia.
About Nutriband Inc.
We are primarily engaged in the development of a portfolio of transdermal pharmaceutical products. Our lead product under development is an abuse-deterrent fentanyl patch incorporating our AVERSA™ abuse-deterrent technology. AVERSA™ technology can be incorporated into any transdermal patch to prevent the abuse, misuse, diversion, and accidental exposure of drugs with abuse potential.
The Company’s website is www.nutriband.com. Any material contained in or derived from the Company’s websites or any other website is not part of this press release.
About Kindeva
At Kindeva, we manufacture more tomorrows for patients worldwide. With best-in-class facilities and comprehensive CDMO services, we offer more than manufacturing—we deliver strategic value. Our global network of 10 manufacturing and R&D sites offer exceptional integrated knowledge and capabilities, including Annex 1-compliant state-of-the-art aseptic fill finish capacity and next-generation sustainable inhalation propellant technology. By combining expertise in injectable, pulmonary, nasal and dermal drug delivery, we help meet the demands of today and deliver the possibilities of tomorrow. Find out more at https://www.kindevadd.com.
Forward-Looking Statements
Certain statements contained in this press release, including, without limitation, statements containing the words ‘’believes,” “anticipates,” “expects” and words of similar import, constitute “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve both known and unknown risks and uncertainties. The Company’s actual results may differ materially from those anticipated in its forward-looking statements as a result of a number of factors, including those including the Company’s ability to develop its proposed abuse-deterrent fentanyl transdermal system and other proposed products, its ability to obtain patent protection for its abuse technology, its ability to obtain the necessary financing to develop products and conduct the necessary clinical testing, its ability to obtain Federal Food and Drug Administration approval to market any product it may develop in the United States and to obtain any other regulatory approval necessary to market any product in other countries, including countries in Europe, its ability to market any product it may develop, its ability to create, sustain, manage or forecast its growth; its ability to attract and retain key personnel; changes in the Company’s business strategy or development plans; competition; business disruptions; adverse publicity and international, national and local general economic and market conditions and risks generally associated with an undercapitalized developing company, as well as the risks contained under “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in the Company’s Form S-1, Forms 10-K’s and Forms 10-Q’s, and the Company’s other filings with the Securities and Exchange Commission. Except as required by applicable law, we undertake no obligation to revise or update any forward-looking statements to reflect any event or circumstance that may arise after the date hereof.
ATHENS, Greece, June 18, 2025 (GLOBE NEWSWIRE) — Euroseas Ltd. (NASDAQ: ESEA, the “Company” or “Euroseas”), an owner and operator of container carrier vessels and provider of seaborne transportation for containerized cargoes, announced today its results for the three-month period ended March 31, 2025 and declared a common stock dividend.
First Quarter 2025 Financial Highlights:
Total net revenues of $56.3 million. Net income of $36.9 million or $5.31 and $5.29 earnings per share basic and diluted, respectively. Adjusted net income1 for the period was $26.2 million or $3.76 per share basic and diluted.
Adjusted EBITDA1 was $37.1 million.
An average of 23.71 vessels were owned and operated during the first quarter of 2025 earning an average time charter equivalent rate of $27,563 per day.
Declared a quarterly dividend of $0.65 per share for the first quarter of 2025 payable on or about July 16, 2025 to shareholders of record on July 9, 2025, as part of the Company’s common stock dividend plan.
On March 17, 2025 the Company completed the spin-off of three of its subsidiaries containing its two older vessels, M/V Aegean Express and M/V Joanna, along with the proceeds from the earlier sale of the vessel M/V Diamantis P, into Euroholdings Ltd. (NASDAQ: EHLD). Beginning on March 18, 2025, Euroholdings Ltd. operates as an independent company.
On May 29, 2025, the Company announced that it has signed an agreement to sell M/V Marcos V, a 6,350 teu intermediate containership built in 2005, to an unaffiliated third party, for $50 million. The vessel is scheduled to be delivered to its buyer in October 2025. The Company is expected to recognize a gain on the sale in excess of $8.50 million, or $1.20 per share.
As of June 18, 2025 we had repurchased 463,074 of our common stock in the open market for a total of about $10.5 million, since the initiation of our share repurchase plan of up to $20 million announced in May 2022.
________________________ 1 Adjusted EBITDA, Adjusted net income and Adjusted earnings per share are not recognized measurements under US GAAP (GAAP) and should not be used in isolation or as a substitute for Euroseas financial results presented in accordance with GAAP. Refer to a subsequent section of the Press Release for the definitions and reconciliation of these measurements to the most directly comparable financial measures calculated and presented in accordance with GAAP.
Aristides Pittas, Chairman and CEO of Euroseas commented: “During the first quarter of 2025, the containership markets showed further strength, with both smaller and larger feeder segments seeing notable rate increases. This positive momentum has continued into the second quarter, with particularly strong gains in the smaller feeder segment. Market strength is also reflected in the secondhand S&P market, where demand for existing tonnage remains firm despite the continued delivery of newbuilds. Reflecting this dynamic, we successfully finalized the sale of one of our intermediate vessels, the M/V Marcos V, to an unaffiliated third party. The market strength is further reflected in our chartering activity resulting in almost 100% charter coverage for 2025 and in excess of 65% for 2026.
“Looking ahead, the containership sector may face notable challenges, primarily due to the high overall orderbook and the possibility that liner companies may resume transits through the Suez Canal. However, elevated geopolitical uncertainty driven by ongoing and escalating tensions between Iran and Israel compounded by uncertainty surrounding the U.S. Administration’s proposed tariffs add another layer of complexity. Specifically, on the supply-side while the orderbook remains high and represents the key challenge for the sector, it is heavily concentrated on larger vessel sizes. In contrast, the feeder and intermediate segments, where our fleet is concentrated, have historically low orderbooks; in addition, due to the higher proportion of older tonnage in these size segments, they are likely to experience a reduction in fleet supply over the coming years. This evolving fleet profile supports the view that, despite the potential risk of cascading from larger vessels, the fundamentals for feeder and intermediate containerships remain favorable.
“On the fleet growth front, we continue to consider ways of further modernizing our fleet. We will be soon retrofitting one more of our secondhand vessels with energy-saving devices. We have further improved our fleet profile by having transferred our two oldest ships to Euroholdings, a spin-off from our company, to pursue a separate independent market and investment strategy. Given our solid liquidity position, our Board has decided to maintain our high yielding quarterly dividend of $0.65 per share. We are also continuing our share buyback program, as our shares are trading at a substantial discount to our net asset value, despite the visibility of our revenues and earnings. As always, we remain committed to identifying attractive investment opportunities that enhance shareholder value and drive sustainable returns.”
Tasos Aslidis, Chief Financial Officer of Euroseas commented: “Our revenues for the first quarter of 2025 are increased by approximately 20% compared to the same period of 2024. This was mainly the result of the increased average number of vessels owned and operated in the first quarter of 2025, compared to the corresponding period of 2024. The Company operated an average of 23.68 vessels, versus 19.60 vessels during the same period last year. Net revenues amounted to $56.3 million for the first quarter of 2025 compared to $46.7 million for the first quarter of 2024.
“Total daily vessel operating expenses, including management fees, general and administrative expenses, but excluding drydocking costs, were $6,676 during the first quarter of 2025 compared $7,276 to the same quarter of last year. This was the result of the lower operating costs of the nine newbuilding vessels delivered during last year and in the first quarter of 2025. In the first quarter of 2024 the Company operated only five of these newbuilding vessels, while the rest were delivered gradually until January 2025.
“Adjusted EBITDA1 during the first quarter of 2025 was $37.1 million compared to $24.6 million achieved in the first quarter of last year.
“As of March 31, 2025, our outstanding bank debt (before deducting the unamortized loan fees) was $244.0 million, versus restricted and unrestricted cash of approximately $95.5 million. As of the same date, our scheduled debt repayments over the next 12 months amounted to about $30.7 million (excluding the unamortized loan fees).”
First Quarter 2025 Results: For the first quarter of 2025, the Company reported total net revenues of $56.3 million representing an 20.6% increase over total net revenues of $46.7 million during the first quarter of 2024. On average, 23.68 vessels were owned and operated during the first quarter of 2025 earning an average time charter equivalent rate of $27,563 per day compared to 19.60 vessels in the same period of 2024 earning on average $27,806 per day. The Company reported a net income for the period of $36.9 million, as compared to a net income of $20.0 million for the first quarter of 2024.
Voyage expenses for the first quarter of 2025 amounted to $0.2 million as compared to voyage expenses of $1.0 million for the same period of 2024. The increased amount of 2024 is mainly attributable to bunkers consumption by three of our vessels (M/V “Synergy Antwerp”, M/V “Synergy Oakland” and M/V “Marcos”) during their drydock period.
Vessel operating expenses for the first quarter of 2025 amounted to $12.3 million as compared to $11.4 million for the same period of 2024. The increased amount is due to the higher number of vessels owned and operated in the first quarter of 2025 compared to the corresponding period of 2024.
Depreciation expense for the first quarter of 2025 amounted to $8.0 million compared to $5.4 million for the same period of 2024 due to the increased number of vessels in the Company’s fleet.
Related party management fees for the first quarter of 2025 increased to $2.0 million from $1.6 million for the same period of 2024 as a result of the higher number of vessels in our fleet and the adjustment for inflation in the daily vessel management fee, effective from January 1, 2025, increasing it from 810 Euros to 840 Euros.
In the first quarter of 2025 two of our vessels completed extensive repairs afloat for a total cost of $1.8 million. In the first quarter of 2024 three of our vessels completed their special survey with drydock for a total cost of $5.6 million.
General and administrative expenses slightly increased to $1.8 million in the first quarter of 2025, as compared to $1.2 million in the first quarter of 2024 due to increased professional fees and increased cost for our stock incentive plan.
Interest and other financing costs for the first quarter of 2025 amounted to $3.9 million. Capitalized interest charged on the cost of our newbuilding program was $0.1 million for the first quarter of 2025. For the same period of 2024 interest and other financing costs amounted $1.8 million and the capitalized interest charged on the cost of our newbuilding program was $1.4 million. This increase is due to the increased amount of debt in the current period compared to the same period of 2024. For the three months ended March 31, 2025 the Company recognized a $0.17 million loss on its interest rate swap contract, comprising a $0.07 million realized gain and a $0.24 million unrealized loss. For the three months ended March 31, 2024 the Company recognized a $0.86 million gain on its interest rate swap contracts, comprising a $0.10 million realized gain and a $0.76 million unrealized gain.
Adjusted EBITDA1 for the first quarter of 2025 was $37.1 million, compared to $24.6 million achieved for the first quarter of 2024, primarily higher revenues due to the higher number of vessels owned and operated.
Basic and diluted earnings per share for the first quarter of 2025 was $5.31 and $5.29, respectively, calculated on 6,958,137 basic and 6,974,994 diluted weighted average number of shares outstanding compared to basic and diluted earnings per share of $2.89 and $2.87, respectively for the first quarter of 2024, calculated on 6,923,331 basic and 6,969,324 diluted weighted average number of shares outstanding.
The adjusted earnings per share for the quarter ended March 31, 2025 would have been $3.76 per share basic and diluted, respectively, compared to adjusted earnings of $2.67 and $2.66 per share basic and diluted, respectively, for the first quarter of 2024. Usually, security analysts include Adjusted Net Income in their determination of published estimates of earnings per share.
Fleet Profile: The Euroseas Ltd. fleet profile as of June 18, 2025 is as follows:
Summary Fleet Data:
(1) Average number of vessels is the number of vessels that constituted the Company’s fleet for the relevant period, as measured by the sum of the number of calendar days each vessel was a part of the Company’s fleet during the period divided by the number of calendar days in that period.
(2) Calendar days. We define calendar days as the total number of days in a period during which each vessel in our fleet was in our possession including off-hire days associated with major repairs, drydockings or special or intermediate surveys or days of vessels in lay-up. Calendar days are an indicator of the size of our fleet over a period and affect both the amount of revenues and the amount of expenses that we record during that period.
(3) The scheduled off-hire days including vessels laid-up, vessels committed for sale or vessels that suffered unrepaired damages, are days associated with scheduled repairs, drydockings or special or intermediate surveys or days of vessels in lay-up, or vessels that were committed for sale or suffered unrepaired damages.
(4) Available days. We define available days as the Calendar days in a period net of scheduled off-hire days as defined above. We use available days to measure the number of days in a period during which vessels were available to generate revenues.
(5) Commercial off-hire days. We define commercial off-hire days as days a vessel is idle without employment.
(6) Operational off-hire days. We define operational off-hire days as days associated with unscheduled repairs or other off-hire time related to the operation of the vessels.
(7) Voyage days. We define voyage days as the total number of days in a period during which each vessel in our fleet was in our possession net of commercial and operational off-hire days. We use voyage days to measure the number of days in a period during which vessels actually generate revenues or are sailing for repositioning purposes.
(8) Fleet utilization. We calculate fleet utilization by dividing the number of our voyage days during a period by the number of our available days during that period. We use fleet utilization to measure a company’s efficiency in finding suitable employment for its vessels and minimizing the amount of days that its vessels are off-hire for reasons such as unscheduled repairs or days waiting to find employment.
(9) Fleet utilization, commercial. We calculate commercial fleet utilization by dividing our available days net of commercial off-hire days during a period by our available days during that period.
(10) Fleet utilization, operational. We calculate operational fleet utilization by dividing our available days net of operational off-hire days during a period by our available days during that period.
(11) Time charter equivalent rate, or TCE, is a measure of the average daily net revenue performance of our vessels. Our method of calculating TCE is determined by dividing time charter revenue and voyage charter revenue, if any, net of voyage expenses by voyage days for the relevant time period. Voyage expenses primarily consist of port, canal and fuel costs that are unique to a particular voyage, which would otherwise be paid by the charterer under a time charter contract, or are related to repositioning the vessel for the next charter. TCE, which is a non-GAAP measure, provides additional meaningful information in conjunction with voyage revenues, the most directly comparable GAAP measure, because it assists our management in making decisions regarding the deployment and use of our vessels and because we believe that it provides useful information to investors regarding our financial performance. TCE is a standard shipping industry performance measure used primarily to compare period-to-period changes in a shipping company’s performance despite changes in the mix of charter types (i.e., spot voyage charters, time charters and bareboat charters) under which the vessels may be employed between the periods. Our definition of TCE may not be comparable to that used by other companies in the shipping industry.
(12) We calculate daily vessel operating expenses, which includes crew costs, provisions, deck and engine stores, lubricating oil, insurance, maintenance and repairs and related party management fees by dividing vessel operating expenses and related party management fees by fleet calendar days for the relevant time period. Drydocking expenses are reported separately.
(13) Daily general and administrative expenses are calculated by us by dividing general and administrative expenses by fleet calendar days for the relevant time period.
(14) Total vessel operating expenses, or TVOE, is a measure of our total expenses associated with operating our vessels. TVOE is the sum of vessel operating expenses, related party management fees and general and administrative expenses; drydocking expenses are not included. Daily TVOE is calculated by dividing TVOE by fleet calendar days for the relevant time period.
(15) Daily drydocking expenses is calculated by us by dividing drydocking expenses by the fleet calendar days for the relevant period, Drydocking expenses include expenses during drydockings that would have been capitalized and amortized under the deferral method. Drydocking expenses could vary substantially from period to period depending on how many vessels underwent drydocking during the period. The Company expenses drydocking expenses as incurred.
Conference Call and Webcast: Today, Wednesday, June 18, 2025 at 09:30 a.m. Eastern Time, the Company’s management will host a conference call and webcast to discuss the results.
Conference Call details: Participants should dial into the call 10 minutes before the scheduled time using the following numbers: 877 405 1226 (US Toll-Free Dial In) or +1 201 689 7823 (US and Standard International Dial In). Please quote “Euroseas” to the operator and/or conference ID 13754421. Click here for additional participant International Toll -Free access numbers.
Alternatively, participants can register for the call using the call me option for a faster connection to join the conference call. You can enter your phone number and let the system call you right away. Click here for the call me option.
Audio Webcast – Slides Presentation: There will be a live and then archived webcast of the conference call and accompanying slides, available on the Company’s website. To listen to the archived audio file, visit our website http://www.euroseas.gr and click on Company Presentations under our Investor Relations page. Participants to the live webcast should register on the website approximately 10 minutes prior to the start of the webcast.
The slide presentation for the first quarter ended March 31, 2025, will also be available in PDF format minutes prior to the conference call and webcast, accessible on the company’s website (www.euroseas.gr) on the webcast page. Participants to the webcast can download the PDF presentation.
Adjusted EBITDA Reconciliation:
Euroseas Ltd. considers Adjusted EBITDA to represent net income before interest and other financing costs, income taxes, depreciation, (gain) / loss on interest rate swap derivative, net, gain on sale of vessel, and amortization of fair value of below market time charters acquired. Adjusted EBITDA does not represent and should not be considered as an alternative to net income, as determined by United States generally accepted accounting principles, or GAAP. Adjusted EBITDA is included herein because it is a basis upon which the Company assesses its financial performance and liquidity position and because the Company believes that this non-GAAP financial measure assists our management and investors by increasing the comparability of our performance from period to period by excluding the potentially disparate effects between periods of financial costs, loss / (gain) on interest rate swaps, gain on sale of vessel, depreciation, and amortization of below market time charters acquired. The Company’s definition of Adjusted EBITDA may not be the same as that used by other companies in the shipping or other industries.
Adjusted net income and Adjusted earnings per share Reconciliation: Euroseas Ltd. considers Adjusted net income to represent net income before unrealized (gain) / loss on derivative, gain on sale of vessel, amortization of below market time charters acquired and vessel depreciation on the portion of the consideration of vessels acquired with attached time charters allocated to below market time charters. Adjusted net income and Adjusted earnings per share are included herein because we believe they assist our management and investors by increasing the comparability of the Company’s fundamental performance from period to period by excluding the potentially disparate effects between periods of the aforementioned items, which may significantly affect results of operations between periods.
Adjusted net income and Adjusted earnings per share do not represent and should not be considered as an alternative to net income or earnings per share, as determined by GAAP. The Company’s definition of Adjusted net income and Adjusted earnings per share may not be the same as that used by other companies in the shipping or other industries. Adjusted net income and Adjusted earnings per share are not adjusted for all non-cash income and expense items that are reflected in our statement of cash flows.
About Euroseas Ltd. Euroseas Ltd. was formed on May 5, 2005 under the laws of the Republic of the Marshall Islands to consolidate the ship owning interests of the Pittas family of Athens, Greece, which has been in the shipping business over the past 140 years. Euroseas trades on the NASDAQ Capital Market under the ticker ESEA.
Euroseas operates in the container shipping market. Euroseas’ operations are managed by Eurobulk Ltd., an ISO 9001:2008 and ISO 14001:2004 certified affiliated ship management company, which is responsible for the day-to-day commercial and technical management and operations of the vessels. Euroseas employs its vessels on spot and period charters and through pool arrangements.
The Company has a fleet of 22 vessels, including 15 Feeder containerships and 7 Intermediate containerships. Euroseas 22 containerships have a cargo capacity of 67,494 teu. After the delivery of two intermediate containership newbuildings in the fourth quarter of 2027, Euroseas’ fleet will consist of 24 vessels with a total carrying capacity of 76,094 teu.
Forward Looking Statement This press release contains forward-looking statements (as defined in Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended) concerning future events and the Company’s growth strategy and measures to implement such strategy; including expected vessel acquisitions and entering into further time charters. Words such as “expects,” “intends,” “plans,” “believes,” “anticipates,” “hopes,” “estimates,” and variations of such words and similar expressions are intended to identify forward-looking statements. Although the Company believes that the expectations reflected in such forward-looking statements are reasonable, no assurance can be given that such expectations will prove to have been correct. These statements involve known and unknown risks and are based upon a number of assumptions and estimates that are inherently subject to significant uncertainties and contingencies, many of which are beyond the control of the Company. Actual results may differ materially from those expressed or implied by such forward-looking statements. Factors that could cause actual results to differ materially include, but are not limited to changes in the demand for containerships, competitive factors in the market in which the Company operates; risks associated with operations outside the United States; and other factors listed from time to time in the Company’s filings with the Securities and Exchange Commission. The Company expressly disclaims any obligations or undertaking to release publicly any updates or revisions to any forward-looking statements contained herein to reflect any change in the Company’s expectations with respect thereto or any change in events, conditions or circumstances on which any statement is based.
Agreement to support future studies investigating the combination of ateganosine and atezolizumab for safe and effective cancer treatments
CHICAGO–(BUSINESS WIRE)– MAIA Biotechnology, Inc. (NYSE American: MAIA), a clinical-stage biopharmaceutical company focused on developing targeted immunotherapies for cancer, today announced its entry into a clinical master supply agreement with Roche for future studies investigating the combination of MAIA’s telomere-targeting agent ateganosine (THIO), sequenced with Roche’s checkpoint inhibitor (CPI), atezolizumab (Tecentriq®), for the treatment of multiple hard-to-treat cancers.
“In preclinical studies, ateganosine was found to be highly synergistic and effective in combination with Roche’s anti-PD-L1 agent atezolizumab,” said MAIA Chairman and CEO Vlad Vitoc, M.D. “We are pleased to partner with world-renowned Roche and we look forward to further strengthening our mission to find safe and effective cancer treatments.”
About Ateganosine
Ateganosine (THIO, 6-thio-dG or 6-thio-2’-deoxyguanosine) is a first-in-class investigational telomere-targeting agent currently in clinical development to evaluate its activity in non-small cell lung cancer (NSCLC). Telomeres, along with the enzyme telomerase, play a fundamental role in the survival of cancer cells and their resistance to current therapies. The modified nucleotide 6-thio-2’-deoxyguanosine induces telomerase-dependent telomeric DNA modification, DNA damage responses, and selective cancer cell death. Ateganosine-damaged telomeric fragments accumulate in cytosolic micronuclei and activate both innate (cGAS/STING) and adaptive (T-cell) immune responses. The sequential treatment with ateganosine followed by PD-(L)1 inhibitors resulted in profound and persistent tumor regression in advanced, in vivo cancer models by induction of cancer type–specific immune memory. Ateganosine is presently developed as a second or later line of treatment for NSCLC for patients that have progressed beyond the standard-of-care regimen of existing checkpoint inhibitors.
About MAIA Biotechnology, Inc.
MAIA is a targeted therapy, immuno-oncology company focused on the development and commercialization of potential first-in-class drugs with novel mechanisms of action that are intended to meaningfully improve and extend the lives of people with cancer. Our lead program is ateganosine (THIO), a potential first-in-class cancer telomere targeting agent in clinical development for the treatment of NSCLC patients with telomerase-positive cancer cells. For more information, please visit www.maiabiotech.com.
Tecentriq® (atezolizumab) is a registered trademark of Genentech, a member of the Roche Group.
Forward-Looking Statements
MAIA cautions that all statements, other than statements of historical facts contained in this press release, are forward-looking statements. Forward-looking statements are subject to known and unknown risks, uncertainties, and other factors that may cause our or our industry’s actual results, levels or activity, performance or achievements to be materially different from those anticipated by such statements. The use of words such as “may,” “might,” “will,” “should,” “could,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “project,” “intend,” “future,” “potential,” or “continue,” and other similar expressions are intended to identify forward-looking statements. However, the absence of these words does not mean that statements are not forward-looking. For example, all statements we make regarding (i) the initiation, timing, cost, progress and results of our preclinical and clinical studies and our research and development programs, (ii) our ability to advance product candidates into, and successfully complete, clinical studies, (iii) the timing or likelihood of regulatory filings and approvals, (iv) our ability to develop, manufacture and commercialize our product candidates and to improve the manufacturing process, (v) the rate and degree of market acceptance of our product candidates, (vi) the size and growth potential of the markets for our product candidates and our ability to serve those markets, and (vii) our expectations regarding our ability to obtain and maintain intellectual property protection for our product candidates, are forward looking. All forward-looking statements are based on current estimates, assumptions and expectations by our management that, although we believe to be reasonable, are inherently uncertain. Any forward-looking statement expressing an expectation or belief as to future events is expressed in good faith and believed to be reasonable at the time such forward-looking statement is made. However, these statements are not guarantees of future events and are subject to risks and uncertainties and other factors beyond our control that may cause actual results to differ materially from those expressed in any forward-looking statement. Any forward-looking statement speaks only as of the date on which it was made. We undertake no obligation to publicly update or revise any forward-looking statement, whether as a result of new information, future events or otherwise, except as required by law. In this release, unless the context requires otherwise, “MAIA,” “Company,” “we,” “our,” and “us” refers to MAIA Biotechnology, Inc. and its subsidiaries.
ATLANTA, June 17, 2025 (GLOBE NEWSWIRE) — DLH Holdings Corp. (NASDAQ: DLHC) (“DLH” or the “Company”), a leading provider of science research and development, systems engineering and integration, and digital transformation and cybersecurity solutions announced today that three DLH solutions developed in collaboration with military health leadership have been named 2025 FORUM Innovation Award winners.
Each year, the FORUM Innovation Awards recognize top IT programs nominated and selected by their peers for pushing the technology envelope, showcasing breakthrough innovation, and rewarding the leadership and teamwork that improve and advance each agency’s mission.
“DLH and our partners in the military health community operate at the leading-edge of scientific discovery and technological innovation,” said Zach Parker, DLH President and CEO. “Each of these award-winning projects demonstrate the life-saving impact that the work of our data scientists, engineers, and health experts has on Warfighter readiness.”
The 2025 FORUM Innovation Award winners are:
Telerobotic Operator Network (TRON) – DHA MRDC TATRC
TRON is a groundbreaking initiative which allows surgeons to operate on patients located far away by combining virtual reality, digital twin, AI, and robotics. With this technology, doctors and medics can remotely provide vital care on wounded Warfighters operating in hazardous conditions that would ordinarily make treatment nearly impossible.
AutoDoc – DHA MRDC TATRC
Collecting accurate, actionable data is central to developing life-saving automated casualty care solutions, but data collection at the point of care typically requires caregivers to stop providing treatment for the sake of documentation. Automating Documentation (“AutoDoc”) delivers a suite of sensors that passively collect accurate and reliable data on patients and medics in challenging operational environments and high stress situations – allowing medics to focus on the vital care they are providing.
Joint Patient Safety Reporting (JPSR) – DHA PEO Medical Systems, DADIO/J-6
Accurate, comprehensive event reporting is crucial for patient safety, but Warfighter health data is often partitioned between the Defense Health Agency (“DHA”) and Department of Veterans Affairs (“VA”). JPSR securely integrates patient health data into a single system for quantitative and comparative data analysis, including customizable analytical tools, reports, and dashboards which allow for at-a-glance monitoring, measuring, and analysis. This unified system gives caregivers the full visibility they need.
“For over twenty years, DLH has joined forces with military partners to drive research and development, including integrating AI/ML technologies, autonomous medical systems, and interoperable telemedicine platforms,” said Mary Dowdall, President, Advanced Mission Solutions. “These awards demonstrate the value of our enduring collaboration and demonstrate our company’s ability to execute at the nexus of science and technology.”
About DLH
DLH (NASDAQ: DLHC), a Russell 2000 company, enhances technology, public health, and cyber security readiness missions through science, technology, cyber, and engineering solutions and services. Our experts solve some of the most complex and critical missions faced by customers today, leveraging digital transformation, artificial intelligence, advanced analytics, cloud-based applications, telehealth systems, and more. With over 2,400 employees dedicated to the idea that “Your Mission is Our Passion,” DLH brings a unique combination of technology, innovation, and world-class expertise to improve lives across the globe. For more information, visit www.DLHcorp.com.
INVESTOR RELATIONS Contact: Chris Witty Phone: 646-438-9385 Email: cwitty@darrowir.com
NEW YORK–(BUSINESS WIRE)– Vince Holding Corp. (NYSE: VNCE) (“VNCE” or the “Company”), a global contemporary retailer, today reported its financial results for the first quarter ended May 3, 2025.
Brendan Hoffman, Chief Executive Officer of VNCE said, “I continue to be encouraged by the strong execution and commitment to excellence I see across our organization, and while we are navigating a challenging environment marked by uncertainty, our first quarter performance was relatively in line with our expectations. As an organization, we quickly pivoted all efforts in the latter portion of the quarter to develop and put into action mitigation plans in light of the evolving tariff policies. In short order we have diversified our supply chain, negotiated with vendors, and leveraged other opportunities to mitigate near-term costs. As we look ahead, we will continue these efforts along with providing customers a high quality product offering and an engaging experience across our channels.”
In this press release, the Company is presenting its financial results in conformity with U.S. generally accepted accounting principles (“GAAP”) as well as on an “adjusted” basis. Adjusted results presented in this press release are non-GAAP financial measures. See “Non-GAAP Financial Measures” below for more information about the Company’s use of non-GAAP financial measures and Exhibit 3 and Exhibit 4 to this press release for a reconciliation of GAAP measures to such non-GAAP measures.
For the first quarter ended May 3, 2025:
Total Company net sales decreased 2.1% to $57.9 million compared to $59.2 million in the first quarter of fiscal 2024. The year-over-year decline was driven by store closures and remodels which negatively impacted the retail store channel in the direct-to-consumer segment.
Gross profit was $29.2 million, or 50.3% of net sales, compared to gross profit of $29.9 million, or 50.6% of net sales, in the first quarter of fiscal 2024. The decrease in gross margin rate was primarily driven by approximately 260 basis points related to higher freight and duty costs, approximately 120 basis points related to wholesale channel mix, and approximately 60 basis points due to higher distribution and handling costs. These factors were partially offset by approximately 330 basis points related to lower product costs and higher pricing and approximately 80 basis points related to lower promotional activity.
Selling, general, and administrative expenses were $33.6 million, or 58.0% of sales, compared to $31.9 million, or 54.0% of sales, in the first quarter of fiscal 2024. The increase in SG&A dollars was primarily driven by higher marketing and advertising expenses, increased legal, information technology and third-party costs as well as increased expenses related to remodels and relocations.
Loss from operations was $4.4 million compared to income from operations of $5.6 million in the same period last year. Excluding the Gain on Sale of Subsidiary (as defined below) in the first quarter of fiscal 2024, Adjusted loss from operations* in the first quarter of fiscal 2024 was $2.0 million.
The income tax provision was $0 for the first quarter of fiscal 2025, as the Company has year-to-date ordinary pre-tax losses for the interim period and is anticipating annual ordinary pre-tax income for the fiscal year. The Company has determined that it is more likely than not that the tax benefit of the year-to-date loss will not be realized in the current or future years and as such, tax provisions for the interim periods should not be recognized until the Company has year-to-date ordinary pre-tax income. The tax provision in the first quarter of fiscal 2025 compares to an income tax benefit of $0.9 million in the same period last year.
Net loss was $4.8 million or $(0.37) per share compared to net income of $4.4 million or $0.35 per share in the same period last year. Excluding the Gain on Sale of Subsidiary, the Adjusted net loss* was $3.3 million or $(0.26) per share in the first quarter of fiscal 2024.
Adjusted EBITDA* was $(3.0) million compared to $(1.5) million in the same period last year.
The Company ended the quarter with 58 company-operated Vince stores, a net decrease of 4 stores since the first quarter of fiscal 2024.
First Quarter Review
Net sales decreased 2.1% to $57.9 million as compared to the first quarter of fiscal 2024.
Wholesale segment sales increased 0.1% to $30.3 million compared to the first quarter of fiscal 2024.
Direct-to-consumer segment sales decreased 4.4% to $27.6 million compared to the first quarter of fiscal 2024.
Income from operations relating to our reportable segments, Vince Wholesale and Vince Direct-to-consumer, was $8.6 million compared to income from operations of $10.1 million in the same period last year.
Net Sales and Operating Results by Segment:
Balance Sheet
At the end of the first quarter of fiscal 2025, total borrowings under the Company’s debt agreements totaled $34.7 million and the Company had $20.4 million of excess availability under its revolving credit facility.
Net inventory at the end of the first quarter of fiscal 2025 was $62.3 million compared to $56.7 million at the end of the first quarter of fiscal 2024.
During the quarter ended May 3, 2025, the Company did not issue shares of common stock under the ATM program. The Company continues to have shares available under the program to exercise with proceeds to be used as sources, along with cash from operations, to fund future growth.
Outlook
For the second quarter of fiscal 2025 the Company expects the following:
Net sales to be approximately flat to down 3% compared to the prior year period.
Operating Income as a percentage of net sales to be approximately (1)% to 1%.
Adjusted EBITDA as a percentage of net sales to be approximately 1% to 4%.
Given the uncertainty related to the potential impact and duration of current tariff policy, the Company is not providing guidance for the full year fiscal 2025.
Strategic Partnership with Authentic Brands Group
On May 25, 2023, the Company announced that it completed the previously announced transaction (the “Authentic Transaction”) with Authentic Brands Group (“Authentic”).
In connection with the Authentic Transaction, VNCE entered into an exclusive, long-term license agreement (the “License Agreement”) with Authentic for usage of the contributed intellectual property for VNCE’s existing business in a manner consistent with the Company’s current wholesale, retail and e-commerce operations. The License Agreement contains an initial ten-year term and eight ten-year renewal options allowing VNCE to renew the agreement.
*Non-GAAP Financial Measures
In addition to reporting financial results in accordance with GAAP, the Company has provided, with respect to the financial results relating to the three months ended May 3, 2025 and May 4, 2024, adjusted EBITDA, which is a non-GAAP measure. Adjusted EBITDA is calculated as earnings before interest, taxes, depreciation and amortization, share-based compensation, capitalized cloud computing amortization, and gain on sale of Rebecca Taylor, Inc. and its wholly owned subsidiary (“Gain on Sale of Subsidiary”). For the three months ended May 4, 2024, the Company has provided adjusted income (loss) from operations, adjusted income (loss) before income taxes and equity in net loss of equity method investment, adjusted income (loss) before equity in net loss of equity method investment, adjusted net income (loss), and adjusted earnings (loss) per share, which are non-GAAP measures, in order to eliminate the effect of the Gain on Sale of Subsidiary.
The Company believes that the presentation of these non-GAAP measures facilitates an understanding of the Company’s continuing operations without the impact associated with the aforementioned items. While these types of events can and do recur periodically, they are excluded from the indicated financial information due to their impact on the comparability of earnings across periods. Non-GAAP financial measures should not be considered in isolation from, or as a substitute for, financial information prepared in accordance with GAAP. A reconciliation of GAAP to non-GAAP results has been provided in Exhibit 3 and Exhibit 4 to this press release.
Conference Call
A conference call to discuss the first quarter results will be held today, June 17, 2025, at 8:30 a.m. ET, hosted by Vince Holding Corp. Chief Executive Officer, Brendan Hoffman, and Chief Financial Officer, Yuji Okumura. During the conference call, the Company may make comments concerning business and financial developments, trends and other business or financial matters. The Company’s comments, as well as other matters discussed during the conference call, may contain or constitute information that has not been previously disclosed.
Those who wish to participate in the call may do so by dialing (833) 470-1428, conference ID 598215. Any interested party will also have the opportunity to access the call via the Internet at http://investors.vince.com/. To listen to the live call, please go to the website at least 15 minutes early to register and download any necessary audio software. For those who cannot listen to the live broadcast, a recording will be available for 12 months after the date of the event. Recordings may be accessed at http://investors.vince.com.
ABOUT VINCE HOLDING CORP.
Vince Holding Corp. is a global retail company that operates the Vince brand women’s and men’s ready to wear business. Vince, established in 2002, is a leading global luxury apparel and accessories brand best known for creating elevated yet understated pieces for every day effortless style. Vince Holding Corp. operates 44 full-price retail stores, 14 outlet stores, and its e-commerce site, as well as through premium wholesale channels globally. Please visit www.vince.com for more information.
Forward-Looking Statements: This document, and any statements incorporated by reference herein contain forward-looking statements under the Private Securities Litigation Reform Act of 1995. Forward-looking statements include the statements under “Outlook” above as well as statements regarding, among other things, our current expectations about possible or assumed future results of operations of the Company and are indicated by words or phrases such as “may,” “will,” “should,” “believe,” “expect,” “seek,” “anticipate,” “intend,” “estimate,” “plan,” “target,” “project,” “forecast,” “envision” and other similar phrases. Although we believe the assumptions and expectations reflected in these forward-looking statements are reasonable, these assumptions and expectations may not prove to be correct and we may not achieve the results or benefits anticipated. These forward-looking statements are not guarantees of actual results, and our actual results may differ materially from those suggested in the forward-looking statements. These forward-looking statements involve a number of risks and uncertainties, some of which are beyond our control, including, without limitation: changes to and unpredictability in the trade policies and tariffs imposed by the U.S. and the governments of other nations; our ability to maintain adequate cash flow from operations or availability under our revolving credit facility to meet our liquidity needs; general economic conditions; restrictions on our operations under our credit facilities; our ability to improve our profitability; our ability to maintain our larger wholesale partners; our ability to accurately forecast customer demand for our products; our ability to maintain the license agreement with ABG Vince, a subsidiary of Authentic Brands Group; ABG Vince’s expansion of the Vince brand into other categories and territories; ABG Vince’s approval rights and other actions; our ability to realize the benefits of our strategic initiatives; the execution of our customer strategy; our ability to make lease payments when due; our ability to open retail stores under favorable lease terms and operate and maintain new and existing retail stores successfully; our operating experience and brand recognition in international markets; our ability to remediate the identified material weakness in our internal control over financial reporting; our ability to comply with domestic and international laws, regulations and orders; increased scrutiny regarding our approach to sustainability matters and environmental, social and governance practices; competition in the apparel and fashion industry; the transition associated with the appointment of new chief executive officer and new chief financial officer; our ability to attract and retain key personnel; seasonal and quarterly variations in our revenue and income; the protection and enforcement of intellectual property rights relating to the Vince brand; our ability to successfully conclude remaining matters following the wind down of the Rebecca Taylor business; the extent of our foreign sourcing; our reliance on independent manufacturers; our ability to ensure the proper operation of the distribution facilities by third-party logistics providers; fluctuations in the price, availability and quality of raw materials; the ethical business and compliance practices of our independent manufacturers; our ability to mitigate system or data security issues, such as cyber or malware attacks, as well as other major system failures; our ability to adopt, optimize and improve our information technology systems, processes and functions; our ability to comply with privacy-related obligations; our ability to submit a required business plan and regain compliance with the New York Stock Exchange (the “NYSE”) Listed Company Manual and maintain a listing of our common stock on the NYSE; our status as a “controlled company”; our status as a “smaller reporting company”; and other factors as set forth from time to time in our Securities and Exchange Commission filings, including those described under “Item 1A—Risk Factors” in our Annual Report on Form 10-K and Quarterly Reports on Form 10-Q. We intend these forward-looking statements to speak only as of the time of this release and do not undertake to update or revise them as more information becomes available, except as required by law.
Shares Expected to Begin Trading on Split-Adjusted Basis on June 20, 2025
LOS ALTOS, Calif., June 17, 2025 (GLOBE NEWSWIRE) — Unicycive Therapeutics, Inc. (NASDAQ: UNCY), a clinical-stage biotechnology company developing therapies for patients with kidney disease, today announced that it will implement a 1-for-10 reverse split of the issued shares of its common stock, effective at 4:01 p.m. Eastern Time on June 18, 2025. The Company’s common stock is expected to begin trading on a split-adjusted basis when the market opens on June 20, 2025, and will continue to trade on The Nasdaq Capital Market under the symbol “UNCY.” The new CUSIP number for the common stock will be 90466Y 202.
The reverse stock split is intended to increase the bid price of the common stock to enable the Company to regain compliance with the $1.00 minimum bid price requirement for continued listing on The Nasdaq Capital Market. The Company’s stockholders authorized the reverse stock split at the Company’s annual meeting of stockholders held on June 9, 2025 and granted the board the authority to determine a final reverse split ratio.
When the reverse stock split becomes effective, every ten (10) shares of the Company’s common stock issued and outstanding or held by the Company in treasury will automatically be combined and reclassified into one (1) share of common stock. No fractional shares will be issued as a result of the reverse stock split. Stockholders who would otherwise be entitled to receive a fractional share will instead automatically have their fractional interests rounded up to the next whole share, after aggregating all the fractional interests of a holder resulting from the reverse stock split. The reverse stock split will affect all stockholders uniformly and will not change any stockholder’s percentage ownership interest or any stockholder’s proportionate voting power, except for immaterial changes that may result from the treatment of fractional shares. The reverse stock split will not change the number of authorized shares of the Company’s common stock or the par value per share of the Company’s common stock.
The reverse stock split will reduce the number of issued and outstanding shares of the Company’s common stock from approximately 126,409,281 to approximately 12,640,929.
As a result of the reverse stock split, proportionate adjustments will be made to the per share exercise prices of, and the number of shares underlying, the Company’s outstanding stock options, as well as to the number of shares available for future awards granted under the Company’s stock incentive plans. In addition, proportionate adjustments will be made to the per share exercise prices of, and the number of shares underlying, outstanding warrants to purchase shares of the Company’s common stock. Further, proportionate adjustments will also be made to the per share conversion price of the Company’s series A and series B preferred stock, pursuant to their respective terms.
The combination of, and reduction in, the issued shares of common stock as a result of the reverse stock split will occur automatically at the effective time of the reverse stock split without any additional action on the part of the Company’s stockholders. The Company’s transfer agent, Pacific Stock Transfer Company, is acting as the exchange agent for the reverse stock split and will send stockholders of record holding their shares electronically in book-entry form a transaction notice indicating the number of shares of common stock held after the reverse stock split. Stockholders who hold their shares through a broker, bank, or other nominee will have their positions adjusted to reflect the reverse stock split, subject to their broker, bank, or other nominee’s particular processes, and are not expected to be required to take any action in connection with the reverse stock split.
Additional information regarding the reverse stock split can be found in the Company’s definitive proxy statement for the annual meeting of stockholders of the Company held on June 9, 2025, which was filed with the U.S. Securities and Exchange Commission on April 30, 2025, a copy of which is available at www.sec.gov and on the Company’s website.
About Unicycive Therapeutics
Unicycive Therapeutics is a biotechnology company developing novel treatments for kidney diseases. Unicycive’s lead investigational treatment is oxylanthanum carbonate, a novel phosphate binding agent currently under review by the U.S. Food and Drug Administration (FDA) for the treatment of hyperphosphatemia in patients with chronic kidney disease who are on dialysis. Unicycive’s second investigational treatment UNI-494 is intended for the treatment of conditions related to acute kidney injury. It has been granted orphan drug designation (ODD) by the FDA for the prevention of Delayed Graft Function (DGF) in kidney transplant patients and has completed a Phase 1 dose-ranging safety study in healthy volunteers. For more information about Unicycive, visit Unicycive.com and follow us on LinkedIn and X. For more information, please visit Unicycive.com and follow us on LinkedIn and X.
Forward-Looking Statements
The Company cautions you that all statements, other than statements of historical facts, contained in this press release, are forward-looking statements. Forward-looking statements, in some cases, can be identified by terms such as “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “design,” “intend,” “expect,” “could,” “plan,” “potential,” “predict,” “seek,” “should,” “would,” “contemplate,” “project,” “target,” “objective,” or the negative version of these words and similar expressions. In this press release, forward-looking statements include, but are not limited to, statements relating to the timing, completion and effect of the reverse stock split and the Company’s ability to regain compliance with Nasdaq’s minimum bid price requirement and continue to have its common stock listed on The Nasdaq Capital Market. Forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause the Company’s actual results, performance or achievements to be materially different from future results, performance or achievements expressed or implied by the forward-looking statements in this press release, including, without limitation, the risk that Nasdaq may not process the reverse stock split on the expected timeline; the risk that after the reverse stock split the closing bid price of the Company’s common stock is not at least $1.00 per share for a minimum of ten consecutive trading sessions; the potential for Nasdaq to suspend trading in or to delist the Company’s common stock. Forward-looking statements are based upon the Company’s current expectations and involve assumptions that may never materialize or may prove to be incorrect. All forward-looking statements are expressly qualified in their entirety by these cautionary statements. For a detailed description of risks and uncertainties the Company faces, you are encouraged to review the documents the Company files with the SEC including the Company’s recent filings on Form 8-K, Form 10-K and Form 10-Q. You are cautioned not to place undue reliance on forward-looking statements, which speak only as of the date on which they were made. The Company undertakes no obligation to update such statements to reflect events that occur or circumstances that exist after the date on which they were made, except as required by law.
Investor Contacts:
Kevin Gardner LifeSci Advisors kgardner@lifesciadvisors.com
Media Contact:
Rachel Visi Real Chemistry redery@realchemistry.com
RICHMOND, Va.–(BUSINESS WIRE)– Lucky Strike Entertainment (NYSE: LUCK), one of the world’s premier Owner/Operators of location-based entertainment, announced that its 2025 Summer Season Pass has exceeded 200,000 members and over $10.3 million in sales through the second week of June—already surpassing the sales of the entire 2024 program, which brought in $8.5 million. This milestone signals a record-breaking start for the popular offering and underscores the early success of the company’s enhanced, guest-first approach behind the Summer Season Pass experience. In addition, Lucky Strike’s four waterparks have sold 32,000 passes for over $3.2 million to date for 2025, highlighting the all weather offerings of the portfolio.
“We’re pleased to see such a strong response to this year’s Summer Season Pass,” said Lucky Strike Entertainment President Lev Ekster. “We’ve listened to our guests and made several strategic optimizations this season—from accessibility to exciting new perks and seasonal surprises. It’s clear those changes are resonating with consumers as we have seen improved and positive same-store sales performance in the portfolio.”
The 2025 Summer Season Pass allows guests to enjoy two games of bowling every day, along with free shoe rental, at over 350 participating Lucky Strike Entertainment locations (under the brands “Lucky Strike”, “Bowlero” and “AMF” among others), all summer long. Guests can choose between Basic and Premium tiers, designed to fit every budget. Premium passholders also enjoy exclusive perks like free arcade credits each visit and food and beverage discounts.
This year’s program marks a thoughtful evolution of the Summer Season Pass with meaningful upgrades seamlessly woven into the guest experience. From a simplified purchasing experience to in-venue perks that surprise and delight, every enhancement was designed with the ultimate goal of offering an incredible value on the company’s best-in-class entertainment offering.
To keep the momentum going, the company has also recently introduced unique offers—like a free Craft Lemonade only for Premium passholders from June 9–15th, giving them a chance to sample the new program before it officially launched to the public on June 16th.
Lucky Strike Entertainment Chief Marketing Officer Katie Warner stated, “We’re looking forward to the rest of the summer with a continued focus on enhancing the experience for passholders through exclusive perks and memorable in-venue moments as we continue to shape the future of this popular and growing program.”
About Lucky Strike Entertainment Lucky Strike Entertainment is one of the world’s premier location-based entertainment platforms. With over 360 locations across North America, Lucky Strike Entertainment provides experiential offerings in bowling, amusements, water parks, and family entertainment centers. The company also owns the Professional Bowlers Association, the major league of bowling and a growing media property that boasts millions of fans around the globe. For more information on Lucky Strike Entertainment, please visit IR.LuckyStrikeEnt.com.
ATHENS, Greece, June 16, 2025 (GLOBE NEWSWIRE) — Euroseas Ltd. (NASDAQ: ESEA), an owner and operator of container carrier vessels and provider of seaborne transportation for containerized cargoes, announced today that it will release its financial results for the first quarter ended March 31, 2025, on June 18, 2025, before market opens in New York.
On the same day, Wednesday, June 18 at 9:30 a.m. Eastern Time, the Company’s management will host a conference call and webcast to discuss the results.
ConferenceCalldetails: Participants should dial into the call 10 minutes before the scheduled time using the following numbers: 877 405 1226 (US Toll-Free Dial In) or +1 201 689 7823 (US and Standard International Dial In). Please quote “Euroseas” to the operator and/or conference ID 13754421. Click here for additional participant International Toll-Free access numbers.
Alternatively, participants can register for the call using the call me option for a faster connection to join the conference call. You can enter your phone number and let the system call you right away. Click here for the call me option.
AudioWebcast‐Slides Presentation: There will be a live and then archived webcast of the conference call and accompanying slides, available on the Company’s website. To listen to the archived audio file, visit our website http://www.euroseas.gr and click on Company Presentations under our Investor Relations page. Participants to the live webcast should register on the website approximately 10 minutes prior to the start of the webcast.
The slide presentation for the first quarter ended March 31, 2025, will also be available in PDF format minutes prior to the conference call and webcast, accessible on the company’s website (www.euroseas.gr) on the webcast page. Participants to the webcast can download the PDF presentation.
AboutEuroseasLtd. Euroseas Ltd. was formed on May 5, 2005 under the laws of the Republic of the Marshall Islands to consolidate the ship owning interests of the Pittas family of Athens, Greece, which has been in the shipping business over the past 140 years. Euroseas trades on the NASDAQ Capital Market under the ticker ESEA. Euroseas operates in the container shipping market. Euroseas’ operations are managed by Eurobulk Ltd., an ISO 9001:2008 and ISO 14001:2004 certified affiliated ship management company, which is responsible for the day-to-day commercial and technical management and operations of the vessels. Euroseas employs its vessels on spot and period charters and through pool arrangements. The Company has a fleet of 22 vessels, including 15 Feeder containerships and 7 Intermediate containerships. Euroseas 22 containerships have a cargo capacity of 67,494 teu. After the delivery of two feeder containership newbuildings in the the fourth quarter of 2027, Euroseas’ fleet will consist of 24 vessels with a total carrying capacity of 76,094 teu.
MALVERN, Pa., June 16, 2025 (GLOBE NEWSWIRE) — Ocugen, Inc. (Ocugen or the Company) (NASDAQ: OCGN), a pioneering biotechnology leader in gene therapies for blindness diseases, today announced that the U.S. Food and Drug Administration (FDA) has cleared the Investigational New Drug (IND) amendment to initiate a Phase 2/3 pivotal confirmatory trial of OCU410ST, a modifier gene therapy candidate being developed for all Stargardt disease (ABCA4-associated retinopathies). The FDA previously granted Rare Pediatric Disease Designation (RPDD) and Orphan Drug Designation for OCU410ST for the treatment of ABCA4-associated retinopathies including Stargardt disease, retinitis pigmentosa 19, and cone-rod dystrophy 3.
“We have had a highly productive and collaborative engagement with the FDA’s Center for Biologics Evaluation and Research (CBER) in establishing the pivotal confirmatory trial for OCU410ST,” said Dr. Shankar Musunuri, Chairman, CEO and Co-Founder of Ocugen. “It’s evident that there is a real sense of urgency by the agency in providing treatment options for patients who currently have nothing available to them. As we initiate the Phase 2/3 registration trial, we are expediting the clinical development of OCU410ST by two to three years and potentially providing an innovative gene therapy to patients desperate for a treatment option.”
Positive data from the Phase 1 GARDian trial for OCU410ST demonstrated:
A favorable safety and tolerability profile with no serious adverse events related to OCU410ST, including no cases of ischemic optic neuropathy, vasculitis, intraocular inflammation, endophthalmitis or choroidal neovascularization and no adverse events of special interest
Considerably slower lesion growth—48% at 12-month follow up in evaluable treated eyes when compared to untreated eyes
Statistically significant (p=0.031) improvement with clinically meaningful, nearly 2-line gain in visual function (BCVA) at 12-month follow-up in evaluable treated eyes when compared to untreated eyes
The Phase 2/3 clinical trial for OCU410ST will enroll 51 participants diagnosed with Stargardt disease. Of these, 34 will receive a one-time subretinal injection of OCU410ST (200 μL at a concentration of 1.5 × 10¹¹ vector genomes/mL) in the eye with poorer visual acuity, while 17 will be assigned to an untreated control group. The primary objective of the trial is to evaluate the reduction in atrophic lesion size. Key secondary endpoints include improvements in best corrected visual acuity (BCVA) and low luminance visual acuity (LLVA), compared to controls. Data from the one-year follow-up will be used to support the company’s Biologics License Application (BLA).
“The initiation of this pivotal Phase 2/3 study represents a significant milestone in our commitment to bringing transformative genetic therapies to individuals affected by Stargardt disease—a progressive and debilitating condition,” said Dr. Huma Qamar, Chief Medical Officer at Ocugen. “The recent RPDD granted by the FDA for this program further underscores the urgent need for innovative treatment options for children living with Stargardt disease. OCU410ST, developed through our proprietary modifier gene therapy platform, is designed to target the underlying biological mechanisms of the disease.”
Approximately 100,000 patients in U.S. and Europe combined and 1 million patients globally live with Stargardt disease. Stargardt and ABCA4-associated retinopathies are genetically complex, involving more than 1,200 known mutations and addressing this condition with traditional gene therapy or gene editing approaches remains highly challenging.
“Stargardt disease represents a significant unmet medical need, particularly among children and young adults,” said Lejla Vajzovic, MD, FASRS, Director of the Duke Surgical Vitreoretinal Fellowship Program and Professor of Ophthalmology, Pediatrics, and Biomedical Engineering with Tenure at Duke University Eye Center. “The Phase 2/3 study of OCU410ST is thoughtfully designed with scientific rigor and a patient-centered focus to evaluate both structural and functional outcomes. We are optimistic that this approach will move us closer to a meaningful therapeutic solution for affected families.”
The OCU410ST Phase 2/3 pivotal confirmatory trial represents a major advancement as Ocugen’s second late-stage clinical program. Ocugen plans to submit a BLA for OCU410ST in 2027 in alignment with its strategic goal of filing three BLAs over the next three years.
About OCU410ST OCU410ST utilizes an AAV delivery platform for the retinal delivery of the RORA (RAR-Related Orphan Receptor A) gene. It represents Ocugen’s modifier gene therapy approach, which is based on Nuclear Hormone Receptor (NHR) RORA that regulates pathophysiological pathways linked to Stargardt disease, such as lipofuscin formation, oxidative stress, complement formation, inflammation, and cell survival networks.
About Stargardt Disease Stargardt disease is a genetic eye disorder that causes retinal degeneration and vision loss. Stargardt disease is the most common form of inherited macular degeneration. The progressive vision loss associated with Stargardt disease is caused by the degeneration of photoreceptor cells in the central portion of the retina called the macula.
Decreased central vision due to loss of photoreceptors in the macula is the hallmark of Stargardt disease. Some peripheral vision is usually preserved. Stargardt disease typically develops during childhood or adolescence, but the age of onset and rate of progression can vary. The retinal pigment epithelium (RPE), a layer of cells supporting photoreceptors, is also affected in people with Stargardt disease.
About Ocugen, Inc. Ocugen, Inc. is a biotechnology company focused on discovering, developing, and commercializing novel gene therapies to address major blindness diseases and offer hope for patients across the globe. We are making an impact on patient’s lives through courageous innovation—forging new scientific paths that harness our unique intellectual and human capital. Our breakthrough modifier gene therapy platform has the potential to address significant unmet medical need for large patient populations through our gene-agnostic approach. Discover more at www.ocugen.com and follow us on X and LinkedIn.
Cautionary Note on Forward-Looking Statements Thispressreleasecontainsforward-lookingstatementswithinthemeaningofThePrivateSecuritiesLitigationReformActof1995,including,butnot limited to, statements regarding qualitative assessments of available data, potential benefits, expectations for ongoing clinical trials, anticipated regulatory filings and anticipated development timelines,whicharesubjecttorisksanduncertainties.Wemay,insomecases,usetermssuchas “predicts,” “believes,” “potential,” “proposed,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Such statements are subject to numerous important factors, risks, and uncertainties that may cause actual events or results to differ materially from our current expectations, including,butnotlimitedto,therisksthatpreliminary,interimandtop-lineclinicaltrialresultsmaynotbeindicativeof,andmaydifferfrom,finalclinical data;the ability of OCU410ST to perform in humans in a manner consistent with nonclinical, preclinical or previous clinical study data;thatunfavorablenewclinicaltrialdatamayemergeinongoingclinicaltrialsorthroughfurtheranalysesofexistingclinicaltrialdata;thatearlier non-clinicalandclinicaldataandtestingofmaynotbepredictiveoftheresultsorsuccessoflaterclinicaltrials;andthatthatclinicaltrialdataare subject to differing interpretations and assessments, including by regulatory authorities.Theseandotherrisksanduncertaintiesaremorefully describedinourperiodicfilingswiththeSecuritiesandExchangeCommission(SEC),includingtheriskfactorsdescribedinthesectionentitled“Risk Factors”inthequarterlyandannualreportsthatwefilewiththeSEC.Anyforward-lookingstatementsthatwemakeinthispressreleasespeakonlyas ofthedateofthispressrelease.Exceptasrequiredbylaw,weassumenoobligationtoupdateforward-lookingstatementscontainedinthispress release whether as a result of new information, future events, or otherwise, after the date of this press release.
TNX-102 SL is a sublingual formulation of cyclobenzaprine designed for transmucosal delivery and durable activity in treating fibromyalgia: FDA PDUFA goal date of August 15, 2025
TNX-102 SL demonstrated statistically significant improvement in the primary endpoint of reduction in fibromyalgia pain in two double-blind randomized placebo-controlled Phase 3 studies
If approved by FDA, TNX-102 SL would become the first member of a new class of non-opioid analgesic drugs for fibromyalgia and the first new drug for treating fibromyalgia in more than 15 years
CHATHAM, N.J., June 16, 2025 (GLOBE NEWSWIRE) — Tonix Pharmaceuticals Holding Corp. (Nasdaq: TNXP) (Tonix or the Company) presented data in a poster presentation at the Annual European Congress of Rheumatology (EULAR) 2025, held June 11-14, 2025, in Barcelona, Spain. A copy of the Company’s poster, titled “Advancing Fibromyalgia Treatment: Transmucosal Sublingual Cyclobenzaprine (TNX-102 SL) Targets Non-restorative Sleep and Provides Sustained Pain Reduction” is available under the Scientific Presentations tab of the Tonix website at www.tonixpharma.com. TNX-102 SL (cyclobenzaprine HCl sublingual tablets) is a non-opioid analgesic designed for daily bedtime dosing with an FDA Prescription Drug User Fee Act (PDUFA) goal date of August 15, 2025.
“Fibromyalgia is a complex and invisible chronic pain condition which drives many patients to be prescribed chronic opioids which are associated with addiction and overdose,” said Seth Lederman, M.D., Chief Executive Officer of Tonix Pharmaceuticals. “To address the chronic symptoms of fibromyalgia, potential therapeutic options must provide durable benefits. TNX-102 SL has shown statistically significant, durable activity (14 weeks) in reducing fibromyalgia pain in two Phase 3 studies. Designed to target the sleep disturbance of fibromyalgia, TNX-102 SL harnesses the therapeutic activity of cyclobenzaprine in part by reducing in the level of the active metabolite norcyclobenzaprine relative to oral cyclobenzaprine. Norcyclobenzaprine is believed to interfere with the durability of oral cyclobenzaprine’s treatment effect in off-label chronic dosing regimens and in a failed double-blind randomized placebo-controlled trial.1 TNX-102 SL now has the potential to be the first new treatment option for fibromyalgia patients in 15 years.”
TNX-102 SL is designed for transmucosal absorption to bypass first-pass hepatic metabolism. The poster presentation shows the day 20 steady state blood levels from a study of nightly TNX-102 SL dosing in which the peak level of cyclobenzaprine exceeds the level of the active metabolite norcyclobenzaprine during sleep time. In contrast, with nightly oral cyclobenzaprine dosing, pharmacokinetic simulations show that norcyclobenzaprine accumulates to higher levels, and the cyclobenzaprine peak level does not exceed the norcyclobenzaprine level during sleep time.
The poster includes data from the RESILIENT Phase 3 study evaluating the efficacy and safety of TNX-102 SL with a primary endpoint of reducing daily pain numeric rating scale scores after 14 weeks of treatment. TNX-102 SL significantly reduced pain and improved clinical outcomes in fibromyalgia patients while demonstrating a favorable tolerability profile. TNX-102 SL employs a novel mechanism targeting the sleep disturbance in fibromyalgia by acting as a potent antagonist at four post-synaptic receptors, each of which is known to regulate sleep.
About Fibromyalgia
Fibromyalgia is a common chronic pain disorder that is understood to result from amplified sensory and pain signaling within the central nervous system, called central sensitization. Brain imaging studies have localized the functional disorder to the brain’s insula and anterior cingulate cortex. Fibromyalgia afflicts more than 10 million adults in the U.S., the majority of whom are women. Symptoms of fibromyalgia include chronic widespread pain, non-restorative sleep, fatigue, and brain fog (or cognitive dysfunction). Other associated symptoms include mood disturbances, including depression, anxiety, headaches and abdominal pain or cramps. Individuals suffering from fibromyalgia often struggle with their daily activities, have impaired quality of life, and frequently are disabled. Physicians and patients report common dissatisfaction with currently marketed products. Fibromyalgia is now recognized as the prototypic nociplastic syndrome. Nociplastic pain is the third primary type of pain in addition to nociceptive pain and neuropathic pain. Many patients present with pain syndromes that are mixtures of the three primary types of pain. Nociplastic syndromes are associated with central and peripheral sensitization. Fibromyalgia can occur without any identifiable precipitating event. However, many fibromyalgia cases follow one or more precipitating event(s) including: post-operative pain, acute or chronic nociceptive or neuropathic pain states; recovery from an infectious illness; a cancer diagnosis or cancer treatment; a metabolic or endocrine stress; or a traumatic event. In the cases of recovery from an infectious illness, fibromyalgia is considered an Infection-Associated Chronic Condition. In addition to fibromyalgia cases associated with other conditions or stressors, the U.S. National Academies of Sciences, Engineering, and Medicine, has concluded that fibromyalgia is a diagnosable condition that can occur after recovery from COVID-19 in the context of Long COVID.
About TNX-102 SL
TNX-102 SL is a centrally acting, non-opioid investigational drug, designed for chronic use. The tablet is a patented sublingual formulation of cyclobenzaprine hydrochloride developed for bedtime dosing for the management of fibromyalgia. Cyclobenzaprine potently binds and acts as an antagonist at four different post-synaptic neuroreceptor subtypes: serotonergic-5-HT2A, adrenergic-α1, histaminergic-H1, and muscarinic-M1-cholinergic receptors. Together, these interactions are believed to target the non-restorative sleep characteristic of fibromyalgia identified by Professor Harvey Moldofsky in 1975. Cyclobenzaprine is not associated with risk of addiction or dependence. The TNX-102 SL tablet is based on a eutectic formulation of cyclobenzaprine HCl and mannitol that provides a stable product which dissolves rapidly and delivers cyclobenzaprine by the transmucosal route efficiently into the bloodstream. The eutectic protects cyclobenzaprine HCl from interacting with the basifying agent that is also part of the formulation and required for efficient transmucosal absorption. Patents based on TNX-102 SL’s eutectic composition and its properties have issued in the U.S., E.U., Japan, China and many other jurisdictions around the world and provide market protection into 2034. The European Patent Office’s Opposition Division maintained Tonix’s European Patent EP 2 968 992 in unamended form after an Opposition was filed against it by a Sandoz subsidiary, Hexal AG. Hexal AG did not appeal that decision. The formulation of TNX-102 SL was designed specifically for sublingual administration and transmucosal absorption for bedtime dosing to target disturbed sleep, while reducing the risk of daytime somnolence. Clinical pharmacokinetic studies indicated that relative to oral cyclobenzaprine, TNX-102 SL results in higher levels of exposure during the first 2 hours after dosing and in deceased levels of the long-lived active metabolite, norcyclobenzaprine in both single dose and multiple dose studies, consistent with bypassing first pass hepatic metabolism. Cyclobenzaprine is a tertiary amine tricyclic and its active metabolite norcyclobenzaprine is a secondary amine tricyclic. At steady state after 20 days of dosing TNX-102 SL, the dynamic peak level of cyclobenzaprine is higher than the background level of norcyclobenzaprine during sleep time. In contrast, after 20 days of dosing oral cyclobenzaprine, the simulated peak level of cyclobenzaprine is lower than the simulated background level of norcyclobenzaprine.
Tonix Pharmaceuticals Holding Corp.*
Tonix is a fully integrated biotechnology company focused on transforming therapies for pain management and vaccines for public health challenges. Tonix’s development portfolio is focused on central nervous system (CNS) disorders. Tonix’s priority is to advance TNX-102 SL, a product candidate for the management of fibromyalgia, for which an NDA was submitted based on two statistically significant Phase 3 studies for the management of fibromyalgia and for which a PDUFA (Prescription Drug User Fee act) goal date of August 15, 2025 has been assigned for a decision on marketing authorization. The FDA has also granted Fast Track designation to TNX-102 SL for the management of fibromyalgia. TNX-102 SL is also being developed to treat acute stress reaction and acute stress disorder under a Physician-Initiated IND at the University of North Carolina in the OASIS study funded by the U.S. Department of Defense (DoD). Tonix’s immunology development portfolio consists of biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is an Fc-modified humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases. Tonix’s infectious disease portfolio includes TNX-801, a vaccine in development for mpox and smallpox, as well as TNX-4200 for which Tonix has a contract with the U.S. DoD’s Defense Threat Reduction Agency (DTRA) for up to $34 million over five years. TNX-4200 is a small molecule broad-spectrum antiviral agent targeting CD45 for the prevention or treatment of infections to improve the medical readiness of military personnel in biological threat environments. Tonix owns and operates a state-of-the art infectious disease research facility in Frederick, Md. Tonix Medicines, our commercial subsidiary, markets Zembrace® SymTouch® (sumatriptan injection) 3 mg and Tosymra® (sumatriptan nasal spray) 10 mg for the treatment of acute migraine with or without aura in adults.
* Tonix’s product development candidates are investigational new drugs or biologics; their efficacy and safety have not been established and have not been approved for any indication.
1Carette S, et al. Arthritis Rheum. 1994;37(1):32-40.
Zembrace SymTouch and Tosymra are registered trademarks of Tonix Medicines. All other marks are property of their respective owners.
This press release and further information about Tonix can be found at www.tonixpharma.com.
Forward Looking Statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimate,” “expect,” and “intend,” among others. These forward-looking statements are based on Tonix’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, risks related to the failure to obtain FDA clearances or approvals and noncompliance with FDA regulations; risks related to the failure to successfully market any of our products; risks related to the timing and progress of clinical development of our product candidates; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payor reimbursement; limited research and development efforts and dependence upon third parties; and substantial competition. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. Tonix does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K for the year ended December 31, 2024, as filed with the Securities and Exchange Commission (the “SEC”) on March 18, 2025, and periodic reports filed with the SEC on or after the date thereof. All of Tonix’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date thereof.
Zembrace® SymTouch® (sumatriptan succinate) injection (Zembrace) and Tosymra® (sumatriptan) nasal spray are prescription medicines used to treat acute migraine headaches with or without aura in adults who have been diagnosed with migraine.
Zembrace and Tosymra are not used to prevent migraines. It is not known if Zembrace or Tosymra are safe and effective in children under 18 years of age.
Important Safety Information
Zembrace and Tosymra can cause serious side effects, including heart attack and other heart problems, which may lead to death. Stop use and get emergency help if you have any signs of a heart attack:
discomfort in the center of your chest that lasts for more than a few minutes or goes away and comes back
severe tightness, pain, pressure, or heaviness in your chest, throat, neck, or jaw
pain or discomfort in your arms, back, neck, jaw or stomach
shortness of breath with or without chest discomfort
breaking out in a cold sweat
nausea or vomiting
feeling lightheaded
Zembrace and Tosymra are not for people with risk factors for heart disease (high blood pressure or cholesterol, smoking, overweight, diabetes, family history of heart disease) unless a heart exam shows no problem.
Do not use Zembrace or Tosymra if you have:
history of heart problems
narrowing of blood vessels to your legs, arms, stomach, or kidney (peripheral vascular disease)
uncontrolled high blood pressure
hemiplegic or basilar migraines. If you are not sure if you have these, ask your provider.
had a stroke, transient ischemic attacks (TIAs), or problems with blood circulation
severe liver problems
taken any of the following medicines in the last 24 hours: almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, ergotamines, or dihydroergotamine. Ask your provider for a list of these medicines if you are not sure.
are taking certain antidepressants, known as monoamine oxidase (MAO)-A inhibitors or it has been 2 weeks or less since you stopped taking a MAO-A inhibitor. Ask your provider for a list of these medicines if you are not sure.
an allergy to sumatriptan or any of the components of Zembrace or Tosymra
Tell your provider about all of your medical conditions and medicines you take, including vitamins and supplements.
Zembrace and Tosymra can cause dizziness, weakness, or drowsiness. If so, do not drive a car, use machinery, or do anything where you need to be alert.
Zembrace and Tosymra may cause serious side effects including:
changes in color or sensation in your fingers and toes
sudden or severe stomach pain, stomach pain after meals, weight loss, nausea or vomiting, constipation or diarrhea, bloody diarrhea, fever
cramping and pain in your legs or hips; feeling of heaviness or tightness in your leg muscles; burning or aching pain in your feet or toes while resting; numbness, tingling, or weakness in your legs; cold feeling or color changes in one or both legs or feet
increased blood pressure including a sudden severe increase even if you have no history of high blood pressure
medication overuse headaches from using migraine medicine for 10 or more days each month. If your headaches get worse, call your provider.
serotonin syndrome, a rare but serious problem that can happen in people using Zembrace or Tosymra, especially when used with anti-depressant medicines called SSRIs or SNRIs. Call your provider right away if you have: mental changes such as seeing things that are not there (hallucinations), agitation, or coma; fast heartbeat; changes in blood pressure; high body temperature; tight muscles; or trouble walking.
hives (itchy bumps); swelling of your tongue, mouth, or throat
seizures even in people who have never had seizures before
The most common side effects of Zembrace and Tosymra include: pain and redness at injection site (Zembrace only); tingling or numbness in your fingers or toes; dizziness; warm, hot, burning feeling to your face (flushing); discomfort or stiffness in your neck; feeling weak, drowsy, or tired; application site (nasal) reactions (Tosymra only) and throat irritation (Tosymra only).
Tell your provider if you have any side effect that bothers you or does not go away. These are not all the possible side effects of Zembrace and Tosymra. For more information, ask your provider.
This is the most important information to know about Zembrace and Tosymra but is not comprehensive. For more information, talk to your provider and read the Patient Information and Instructions for Use. You can also visit https://www.tonixpharma.com or call 1-888-869-7633.
You are encouraged to report adverse effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088.





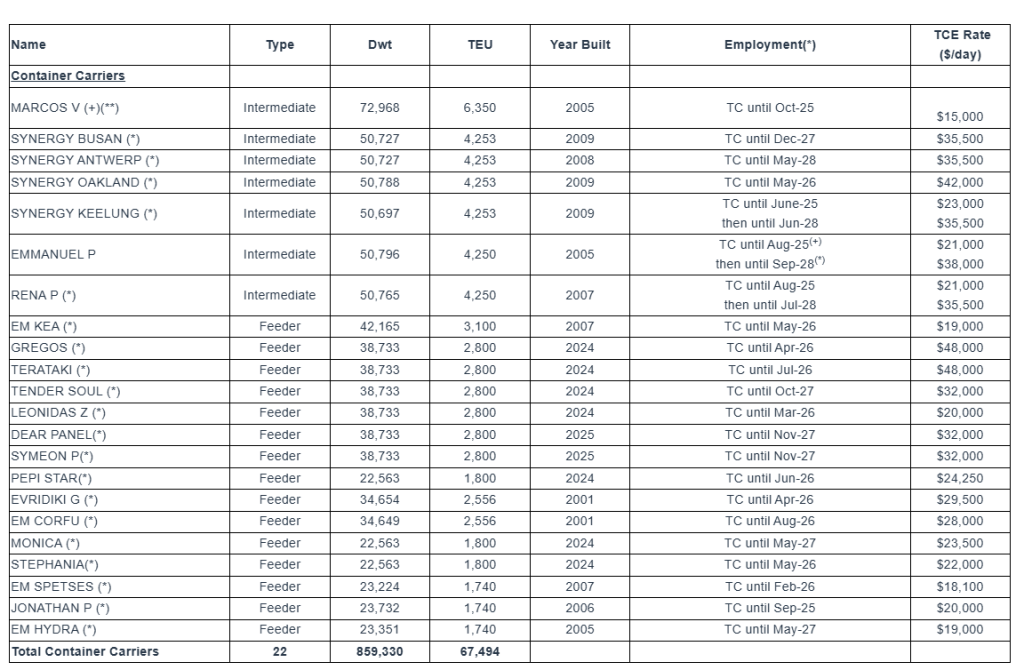
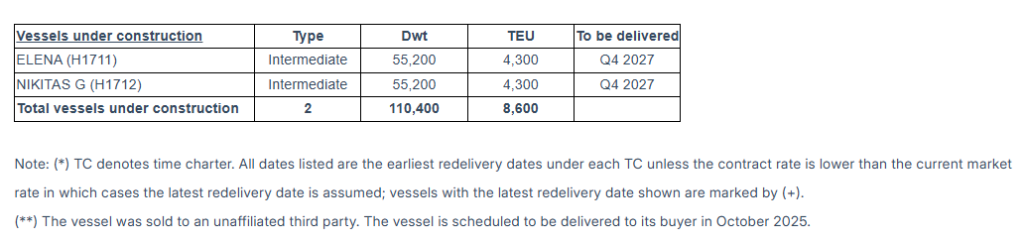
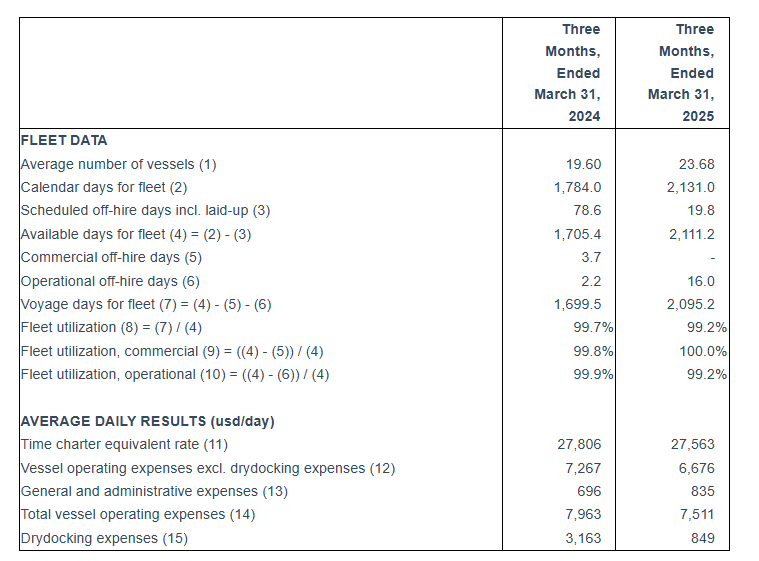
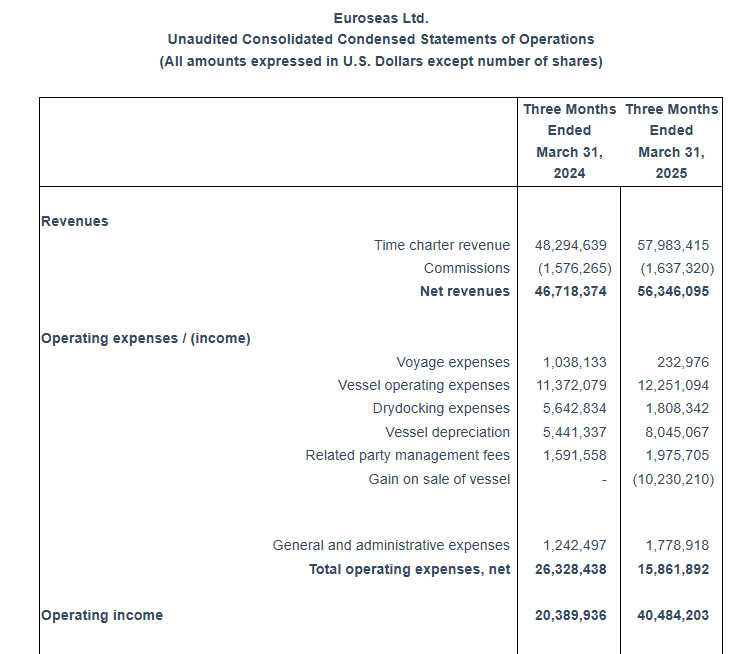
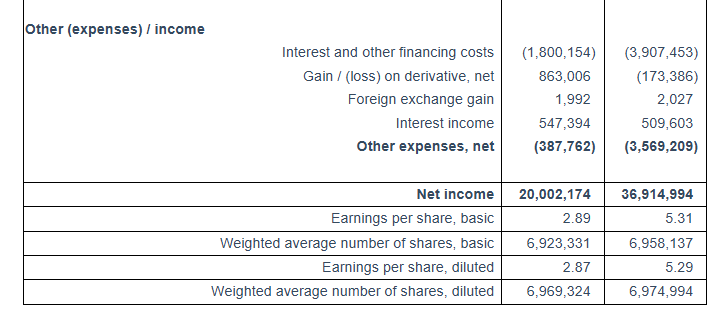


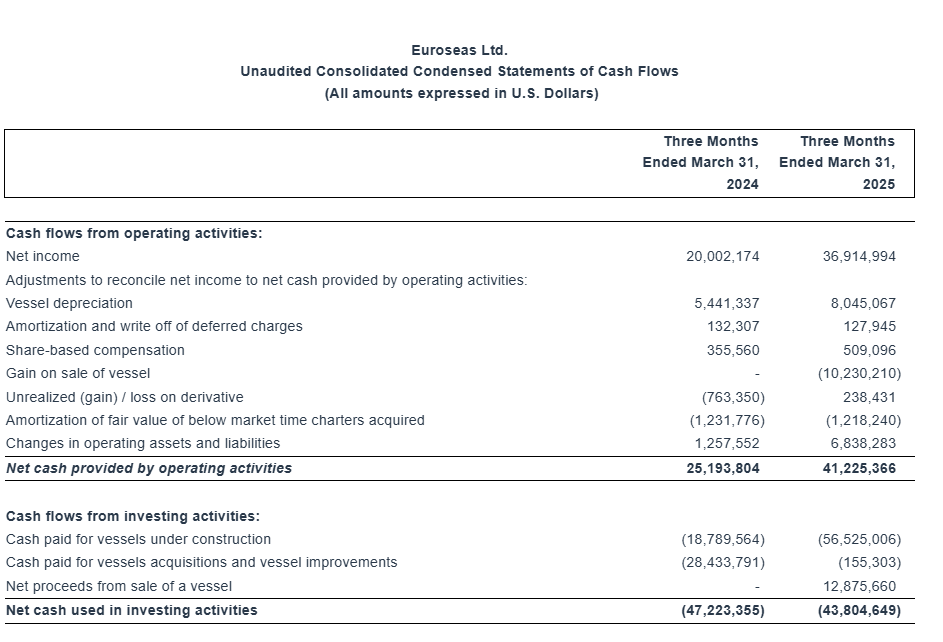
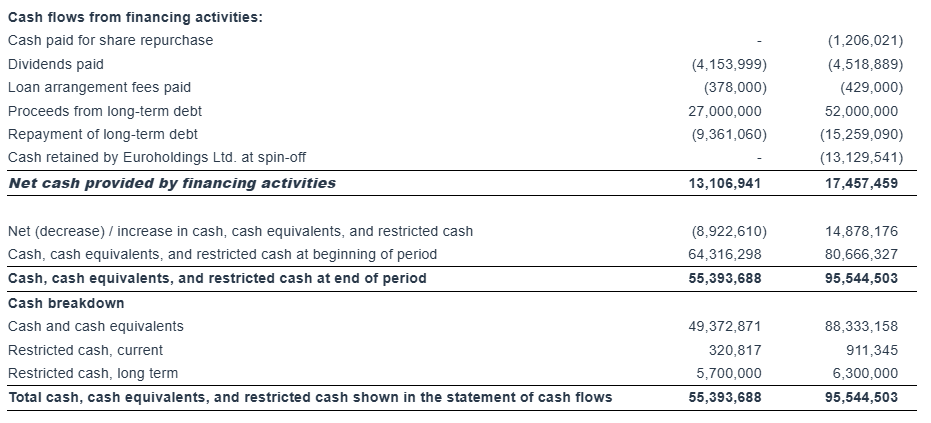

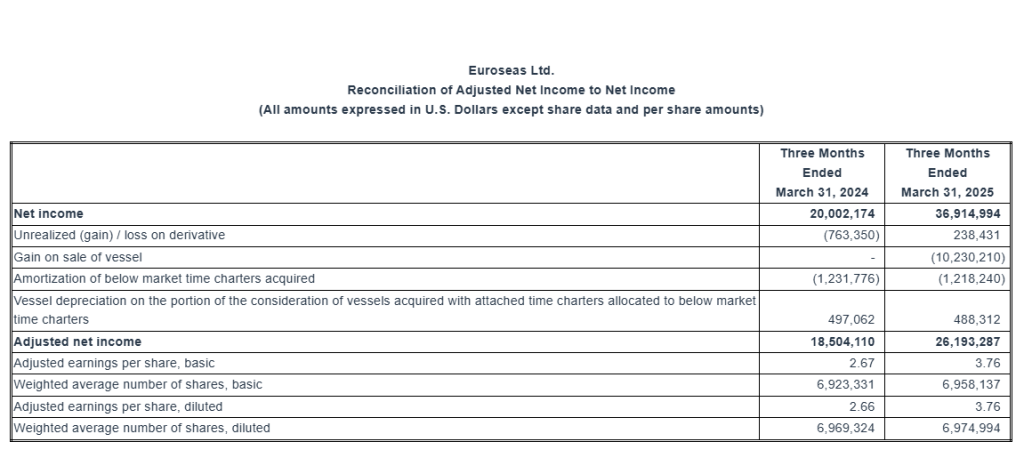



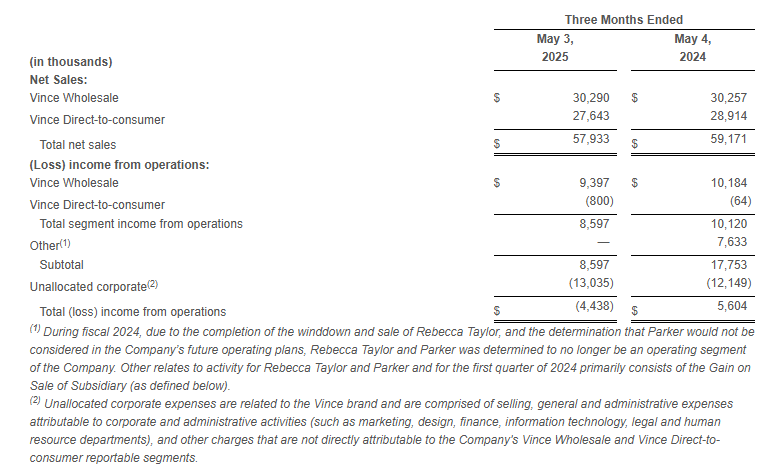
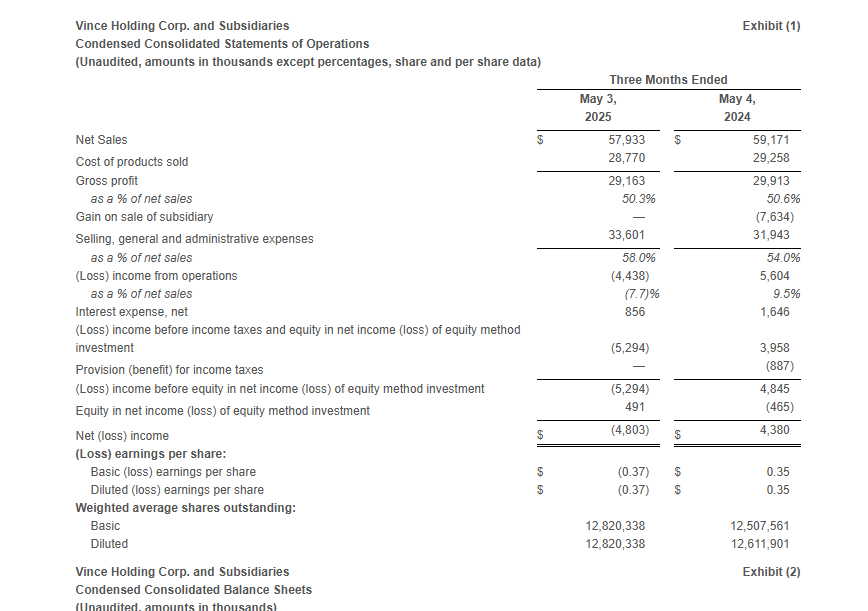
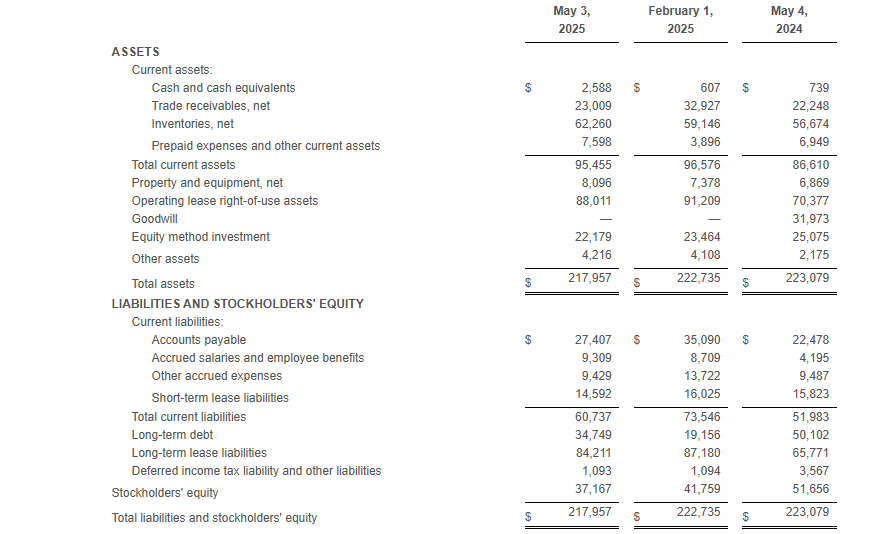
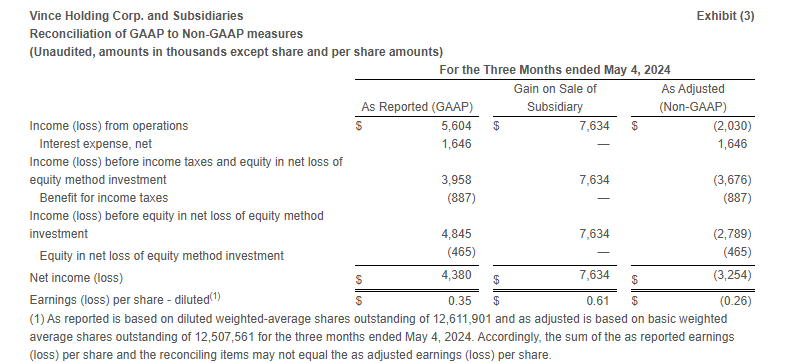




{kind=link}
{kind=link}
{kind=link}